Clear Sky Science · ja
RAD23Aの低下は寿命を延ばし、TDP-43タンパク質病モデルマウスの病理を軽減する
この研究が家族と患者にとって重要な理由
筋萎縮性側索硬化症(ALS)や前頭側頭型認知症(FTD)を含む多くの形の認知症や運動ニューロン疾患では、神経細胞内のタンパク質が誤って折りたたまれて凝集し、ゆっくりとニューロンを毒することが関与しています。主要な原因の一つはTDP-43と呼ばれるタンパク質で、通常はRNAの管理を助けますが、凝集すると有害になります。本研究は希望に満ちた問いを投げかけます:損傷タンパク質の扱われ方を制御する別のタンパク質、RAD23Aの量を下げることで神経細胞をより耐性のある状態にできるか?著者らはマウスでRAD23Aを減らすと寿命が延び、運動性が改善し、TDP-43駆動の疾患モデルにおける脳の損傷が軽減されることを示し、新たな治療戦略の可能性を示唆しています。
病んだニューロン内のタンパク質交通渋滞
神経変性疾患では、しばしば誤った折りたたみタンパク質の堆積が見られ、細胞の廃棄機構がそれを除去できなくなります。ALSやFTDでは、TDP-43が核外へ移動して粘着性の塊を作り、通常はプロテアソームへ導くシグナルであるユビキチンで強く標識されます。RAD23Aはユビキチン標識された貨物をプロテアソームへ運ぶいくつかの「シャトル」タンパク質の一つです。ところが線虫や培養ニューロンでの以前の研究は、RAD23様タンパク質を失うことがTDP-43による損傷からの保護につながる可能性を示唆しており、この矛盾を本研究は哺乳類の生きた脳で検証しようとしました。
TDP-43マウスモデルでのRAD23A抑制
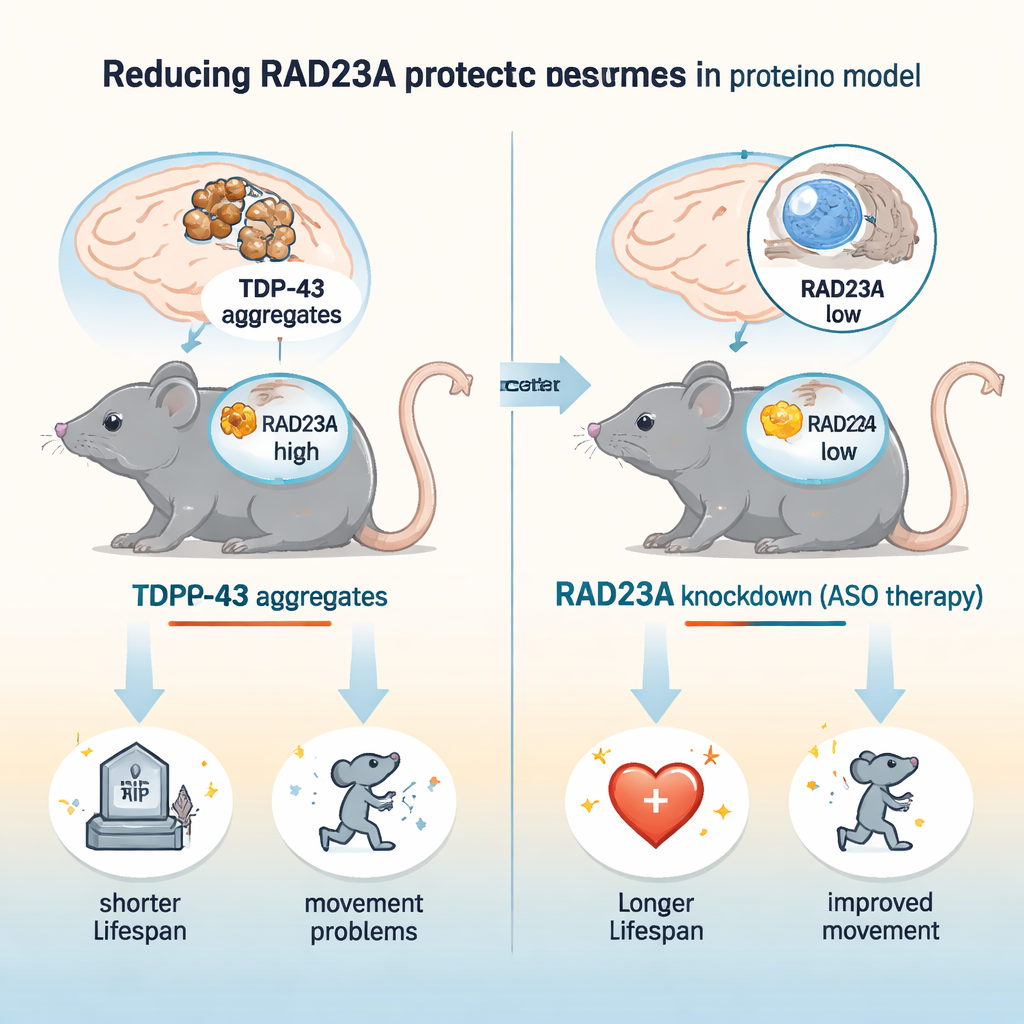
研究者らはTAR4/4と呼ばれる確立されたマウスモデルを用いました。このモデルはニューロンでヒトTDP-43を過剰発現し、運動障害、脊柱湾曲、振戦、早期死亡を示し、ALS/FTDの主要な特徴を再現します。彼らは新生マウスにRad23aのRNAを減少させるアンチセンスオリゴヌクレオチド(ASO)を注入する方法と、Rad23aの遺伝子欠失を持つ系統と交配する方法の二通りでRAD23Aを低下させました。単回のASO処置で脳および脊髄のRAD23Aレベルは約4分の1に減少しました。これらのTDP-43マウスでは、RAD23Aノックダウンにより寿命が約50%延び、歩行障害、振戦、脊柱湾曲、後肢抱え込みの発症と重症度が遅延しました。興味深いことに、RAD23Aの完全な遺伝学的欠失はさらなる利益をもたらさず、部分的な抑制が最適であり、長期的な完全欠失は代償的変化を引き起こす可能性があることを示唆しています。
炎症の軽減、タンパク質処理の改善、そして安定したゲノム
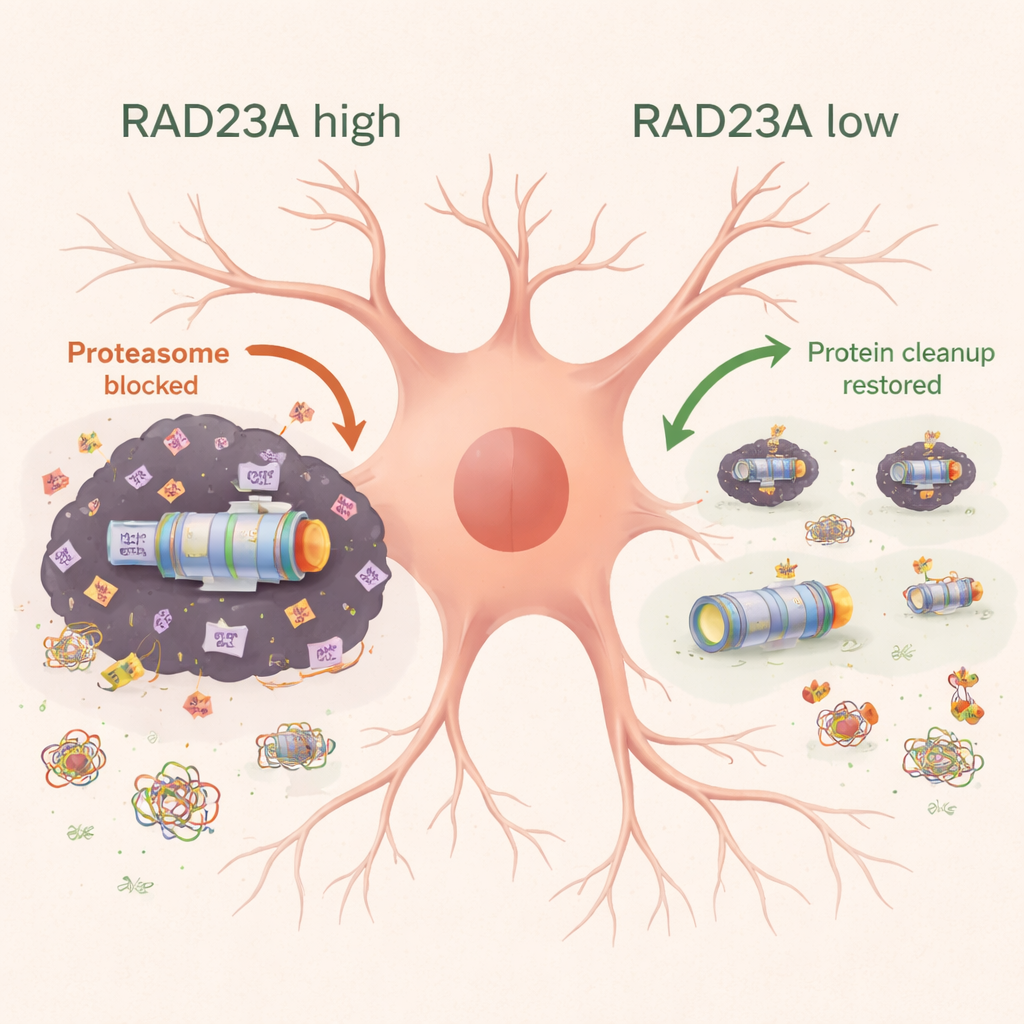
運動皮質の顕微鏡解析では、TDP-43マウスはニューロンの喪失と、星状膠細胞やミクログリアといった脳の支持・免疫細胞の強い活性化を示しました。RAD23Aの低下はニューロン数を保ち、炎症や細胞死のマーカーを低下させました。生化学的解析は、TDP-43の過剰発現がユビキチン標識され、界面活性剤に不溶なタンパク質を細胞に流入させ、プロテアソームサブユニットをこれらの凝集体に引き込み、損傷タンパク質を除去する細胞能を弱めることを明らかにしました。RAD23Aの低下はユビキチン化タンパク質の総負荷を減らし、より多くのプロテアソームを可溶で機能するプールに留め、いくつかのプロテアソーム活性を正常に近づけました。同時に、RAD23Aノックダウンは総TDP-43と凝集型TDP-43の両方(特に毒性の高い25キロダルトン断片を含む)を減少させ、TDP-43を細胞質から核へと再配置しました。ゲノムワイドなRNAシーケンシングは、TDP-43によって引き起こされた何千もの遺伝子発現変化がRAD23A低下によって部分的に逆転されることを示し、特にニューロン機能、ミトコンドリアのエネルギー産生、アグレファジーなどの凝集体除去経路に関与する遺伝子で顕著でした。
不可溶性プロテオームの再編成
通常の界面活性剤に抵抗する頑固な凝集体を詳しく調べるため、研究チームは重同位体質量分析を用いてマウス皮質の不溶性画分に捕らえられたタンパク質をカタログ化しました。ヒトTDP-43の発現はプロテアソーム成分、細胞骨格および輸送タンパク質、その他の細胞機構を引き寄せました。RAD23Aがノックダウンされると、この不溶性プロテオームの全体的な組成は変化しました:プロテアソームや輸送関連タンパク質が隔離される割合は減少し、一方でリボソームやストレス関連タンパク質が凝集体に増加しました。注目すべきは、この再編成が単にRNAレベルでの変化を反映するものではなく、RAD23Aは主に既存タンパク質の可溶性と凝集性の間の振り分けに影響を与え、各タンパク質の産生量そのものを大きく変えるわけではないことを示唆している点です。
将来の治療法にとっての意義
これらの発見は、RAD23Aがストレス下のニューロンにおけるタンパク質品質管理の強力な調整因子であることを描き出します。TDP-43駆動モデルでRAD23Aを部分的に低下させることで、著者らは有害なタンパク質凝集を減らし、タンパク質廃棄機構の活性を回復させ、有害な遺伝子発現変化を抑え、脳の炎症を制限し、寿命と運動機能を延長することができました。異常なTDP-43の蓄積は遺伝性および散発性を問わずALS、FTD、関連疾患に広く見られるため、RAD23Aをヒト向けのアンチセンス薬で標的にすることは、必須タンパク質であるTDP-43自体を直接遮断することなくニューロンを保護する手段を提供するかもしれません。ほかのモデルやヒトでの検証が多く残されているものの、本研究は神経変性の共通経路に対する有望な新たな介入点としてRAD23Aを特定しています。
引用: Guo, X., Prajapati, R.S., Chun, J. et al. Reduction of RAD23A extends lifespan and mitigates pathology in a mouse model of TDP-43 proteinopathy. Nat Commun 17, 1820 (2026). https://doi.org/10.1038/s41467-025-65104-4
キーワード: TDP-43, ALS, タンパク質凝集, プロテアソーム, アンチセンス療法