Clear Sky Science · it
Docking competitivo guidato dall’IA per screening virtuale e predizione dell’efficacia dei composti
Ricerche più intelligenti per nuovi farmaci
Trovare nuovi farmaci è un po’ come cercare un ago in un pagliaio composto da milioni di molecole. Questo studio mostra come i progressi recenti dell’intelligenza artificiale possano rendere quella ricerca più veloce ed economica aiutando gli scienziati a prevedere quali molecole hanno maggiori probabilità di aderire a una proteina legata alla malattia e funzionare effettivamente come farmaci. Anziché testare una sostanza chimica alla volta in laboratorio, gli autori usano modelli di IA per far competere virtualmente le molecole e lasciare che i vincitori emergano in cima alla lista.
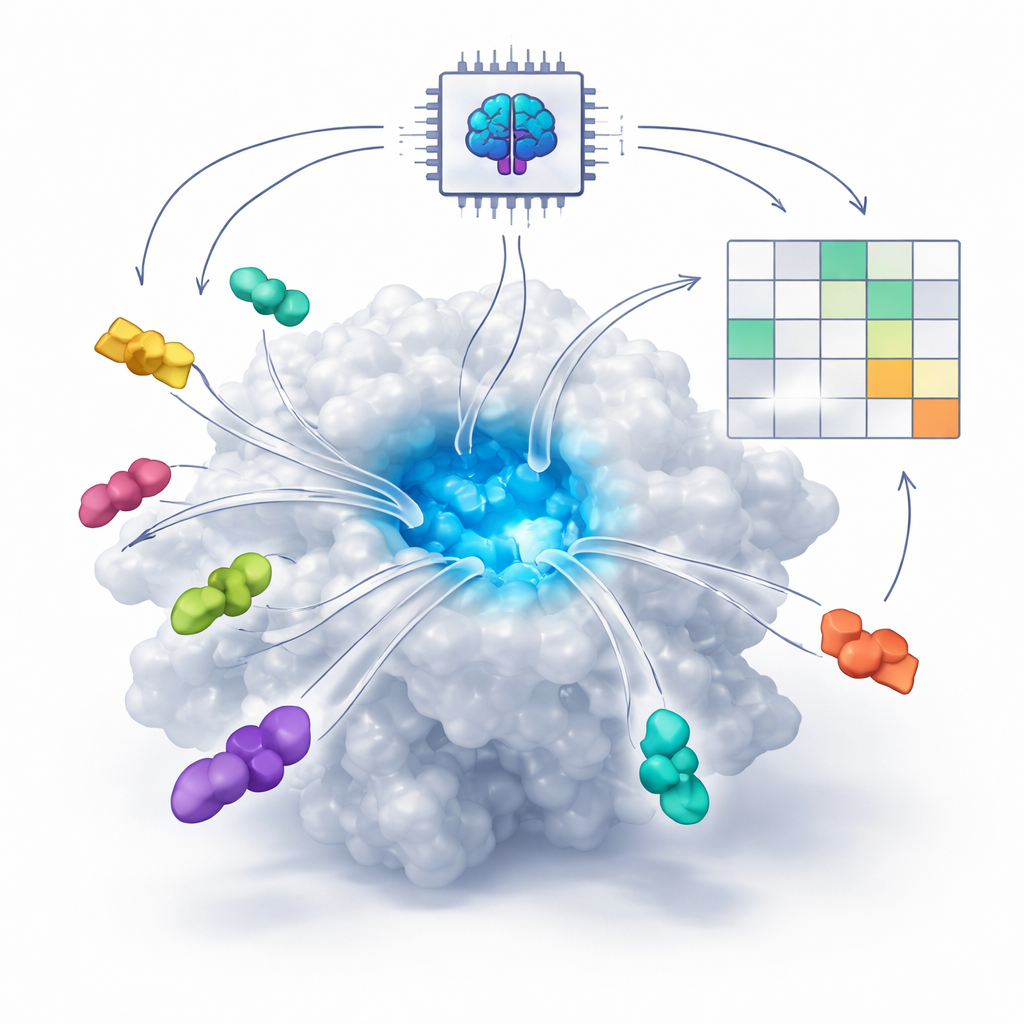
Come l’IA impara a riconoscere l’incastro molecolare a chiave e serratura
Molti farmaci moderni agiscono incastrandosi in piccole tasche sulle proteine, proprio come una chiave entra in una serratura. Tradizionalmente, i programmi informatici cercavano di prevedere questo incastro usando equazioni fisiche che stimano le forze tra atomi. Negli ultimi anni, tuttavia, nuovi sistemi di IA basati su diffusione per co-folding—come AlphaFold3 e Boltz—hanno imparato da un vasto numero di strutture proteina–molecola note. Questi sistemi possono ora “immaginare” come una proteina e un potenziale farmaco possano ripiegarsi insieme in tre dimensioni, anche quando non esiste una struttura sperimentale. La domanda centrale che gli autori affrontano è se questi strumenti IA possano fare più che disegnare immagini plausibili: possono anche distinguere i buoni farmaci dai cattivi?
Leganti reali vs. imitatrici
Il gruppo ha prima testato 16 proteine ben studiate più un enzima batterico più complesso chiamato DNA girasi. Per ciascuna proteina, hanno chiesto ai modelli di IA di collocare sia inibitori attivi noti sia un insieme di molecole “off-target” non correlate nello stesso sito di legame. Invece di fidarsi di una singola previsione, hanno osservato quanto coerentemente l’IA posizionasse ogni molecola attraverso molte esecuzioni. I veri inibitori tendevano a tornare nello stesso punto e nella stessa orientazione ripetutamente, raggruppandosi entro poche trillionesime di metro l’uno dall’altro. Le molecole inattive vagavano più ampiamente e spesso si trovavano più lontano dalla tasca. Questa idea semplice—convergenza delle pose—si è rivelata un segnale forte che un composto si adatta davvero al suo bersaglio proteico.
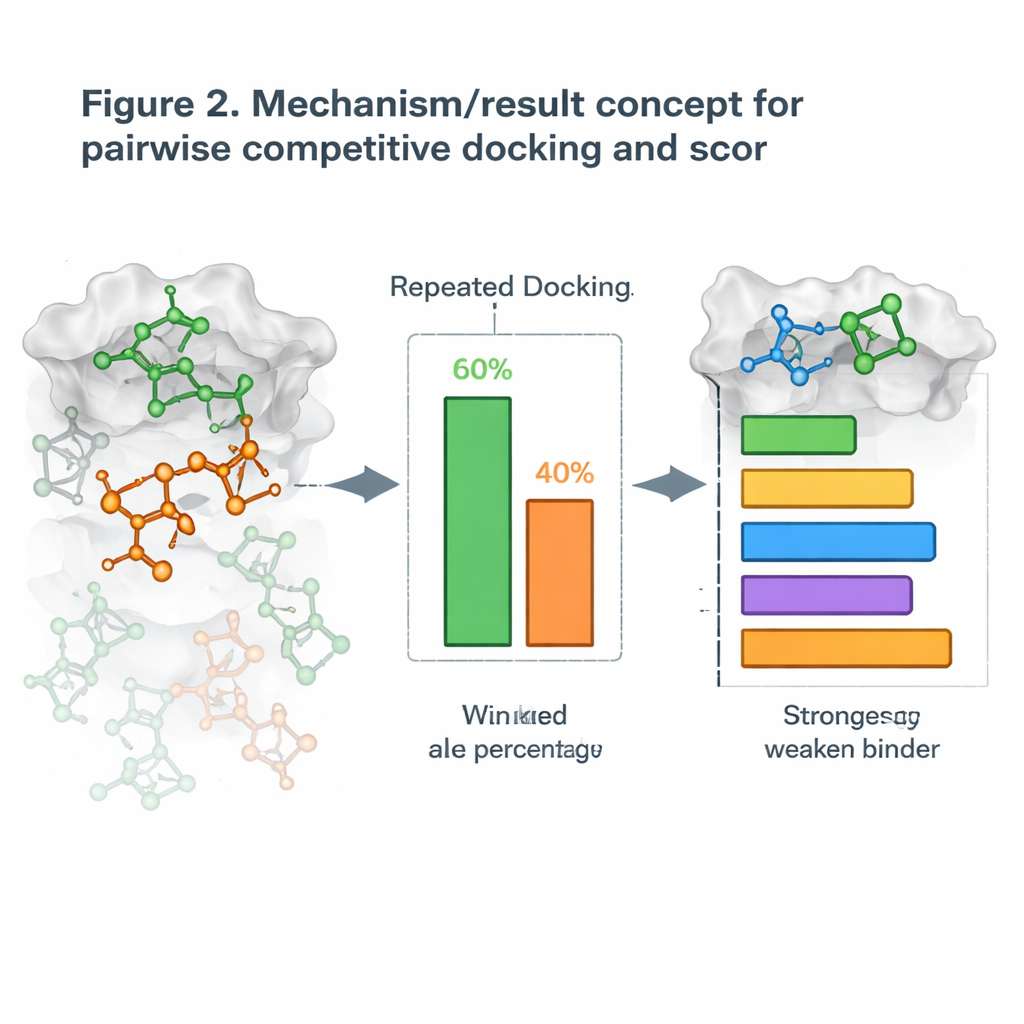
Trasformare il docking in una competizione testa a testa
Partendo da ciò, gli autori hanno introdotto una nuova strategia che chiamano docking competitivo a coppie. Anziché effettuare il docking di una molecola per volta, affiancano due candidati con la proteina e li lasciano “competere” per la stessa tasca. Dopo molte esecuzioni ripetute, la molecola che occupa il sito più frequentemente è dichiarata vincitrice di quell’abbinamento. Eseguendo tutte le possibili coppie, costruiscono una tabella di vittorie-sconfitte e calcolano un punteggio di Docking Competitivo per ogni molecola, simile al ranking dei giocatori in un torneo all’italiana. Quando questi punteggi sono stati confrontati con misure sperimentali reali di quanto fortemente le molecole bloccano i loro bersagli, le classifiche spesso coincidevano bene, con alcuni sistemi proteici che mostravano un accordo quasi perfetto.
Dallo screening virtuale alla progettazione di antibiotici migliori
La DNA girasi, un enzima essenziale per i batteri, è servita come caso di prova dettagliato. Questa proteina presenta diverse tasche per farmaci bersagliate da classi diverse di antibiotici, incluse le ampiamente usate fluorochinoloni. I modelli di IA sono stati in grado, nella maggior parte dei casi, di collocare ciascuna classe di farmaci nella sua tasca corretta, e i punteggi di docking competitivo seguivano grossolanamente le loro potenze misurate. Gli autori hanno quindi scalato il metodo a uno screening virtuale di oltre 3.000 farmaci approvati, chiedendo quali molecole competessero meglio per il sito dei fluorochinoloni. La loro strategia in due fasi—prima usando la competizione “tutti-insieme” per selezionare i probabili vincitori, poi filtrando in base a quanto strettamente si raggruppavano nella tasca—ha arricchito notevolmente i veri fluorochinoloni scartando i candidati più deboli. Infine, hanno usato un generatore di molecole guidato dall’IA per proporre nuove strutture simili ai fluorochinoloni e hanno applicato il docking competitivo per trovare un piccolo numero con legami previsti ancora migliori e proprietà farmacologiche accettabili.
Promesse, limiti e cosa significa per i pazienti
Lo studio mostra che i modelli moderni di IA possono fare più che rappresentare strutture plausibili proteina–farmaco: se eseguiti in un quadro competitivo, possono aiutare a classificare i composti in modo che spesso rispecchia i dati sperimentali reali. Questo non sostituisce il lavoro di laboratorio—le prestazioni dipendono ancora fortemente dalla proteina in questione, alcune tasche sono previste erroneamente e i modelli di IA possono fallire per molecole molto grandi o insolite. Ma con il miglioramento di questi modelli e dei dati di addestramento, approcci come il docking competitivo a coppie potrebbero rendere la scoperta precoce di farmaci molto più efficiente. Per i pazienti, ciò potrebbe infine tradursi in uno sviluppo più rapido di terapie mirate, inclusi nuovi antibiotici capaci di tenere il passo con batteri resistenti.
Citazione: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Parole chiave: scoperta di farmaci con IA, screening virtuale, docking molecolare, legame proteina-ligando, progettazione di antibiotici