Clear Sky Science · it
Calcolo scientifico a basso consumo energetico usando serbatoi chimici
Perché trasformare la chimica in calcolo è importante
I supercomputer moderni consumano quantità enormi di elettricità per simulare il clima, progettare nuovi farmaci o addestrare l’intelligenza artificiale. Avvicinandoci ai limiti fisici dei chip tradizionali, estrarre più prestazioni per watt diventa sempre più difficile e costoso. Questo articolo esplora una strada radicalmente diversa: usare reazioni chimiche reali come motore del calcolo scientifico. Trattando le molecole e le loro interazioni come gli ingranaggi di un computer, gli autori delineano come macchine future potrebbero risolvere equazioni complesse con molta meno energia rispetto all’hardware digitale odierno.
Dalle cellule viventi alle calcolatrici chimiche
Le cellule viventi sono risolutrici di problemi per eccellenza. Gestiscono continuamente migliaia di reazioni per adattarsi, crescere e sopravvivere, consumando al contempo quantità di energia sorprendentemente ridotte. Al centro di questo comportamento ci sono le reti di reazione chimica—reazioni interconnesse i cui tassi e concentrazioni cambiano nel tempo. Queste reti possono essere descritte da equazioni differenziali ordinarie, lo stesso linguaggio matematico usato per modellare tutto, dalle epidemie ai flussi turbolenti. L’intuizione alla base di questo lavoro è che, poiché la chimica già segue queste equazioni, potremmo sfruttarla direttamente per eseguire i calcoli che oggi gli scienziati fanno sui chip di silicio.
Come le equazioni diventano reti di reazioni
Gli autori presentano ChemComp, un framework software che prende un sistema di equazioni differenziali e lo converte sistematicamente in una rete astratta di reazioni. ChemComp utilizza tecnologie moderne di compilazione per scomporre un problema matematico in pattern che possono essere rappresentati da reazioni idealizzate, poi li organizza in una rete con specie, connessioni e costanti di reazione ben definite. Queste reazioni astratte non corrispondono ancora a molecole reali, ma costituiscono un progetto per un computer chimico. Il framework può quindi cercare nei database di reazioni biochimiche per trovare motivi reali di reazione che si comportino in modo simile, privilegiando opzioni pratiche, sicure e potenzialmente a basso consumo energetico in ambiente di laboratorio.
Lasciare che un serbatoio chimico faccia il lavoro pesante
Per mettere alla prova l’idea, il team si concentra su uno stile di apprendimento automatico chiamato reservoir computing. Qui, un sistema dinamico fisso trasforma un segnale di ingresso in un ricco e intricato schema di attività interna, e viene addestrato solo un semplice strato di lettura per produrre l’output desiderato. Nella versione di ChemComp, il reservoir è un insieme di reazioni in un recipiente ben mescolato; le concentrazioni variabili delle sostanze chimiche costituiscono gli stati interni. Gli autori compilano un classico sistema a due variabili noto come modello Sel’kov–Schnakenberg—originariamente usato per studiare oscillazioni nel metabolismo—in reti di reazione candidate. Simulano quindi come queste reti rispondono nel tempo quando vengono alimentate da flussi di sostanze chimiche dentro e fuori dal recipiente, e usano una regressione lineare di base per combinare le tracce di concentrazione in un’approssimazione della soluzione obiettivo.
Testare reti chimiche semplici e più ricche
I ricercatori confrontano due reservoir candidati: uno con solo due specie chimiche e due reazioni, e un altro con cinque specie e cinque reazioni. Entrambe le reti ricevono concentrazioni iniziali e tassi di flusso adeguati, poi vengono simulate mentre operano. Anche il sistema più piccolo è in grado di riprodurre grossolanamente il comportamento oscillatorio delle equazioni target, ma la rete più grande ottiene risultati nettamente migliori, riducendo l’errore sia durante l’addestramento sia nei test. Scansionando diverse concentrazioni iniziali e costanti di velocità di reazione, gli autori mappano le regioni in cui il sistema chimico corrisponde più da vicino alla dinamica desiderata. Ogni reazione agisce in pratica come una funzione base in un problema di adattamento di curve: più reazioni variate sono disponibili, più è semplice approssimare comportamenti complessi, al prezzo di una maggiore complessità del sistema.
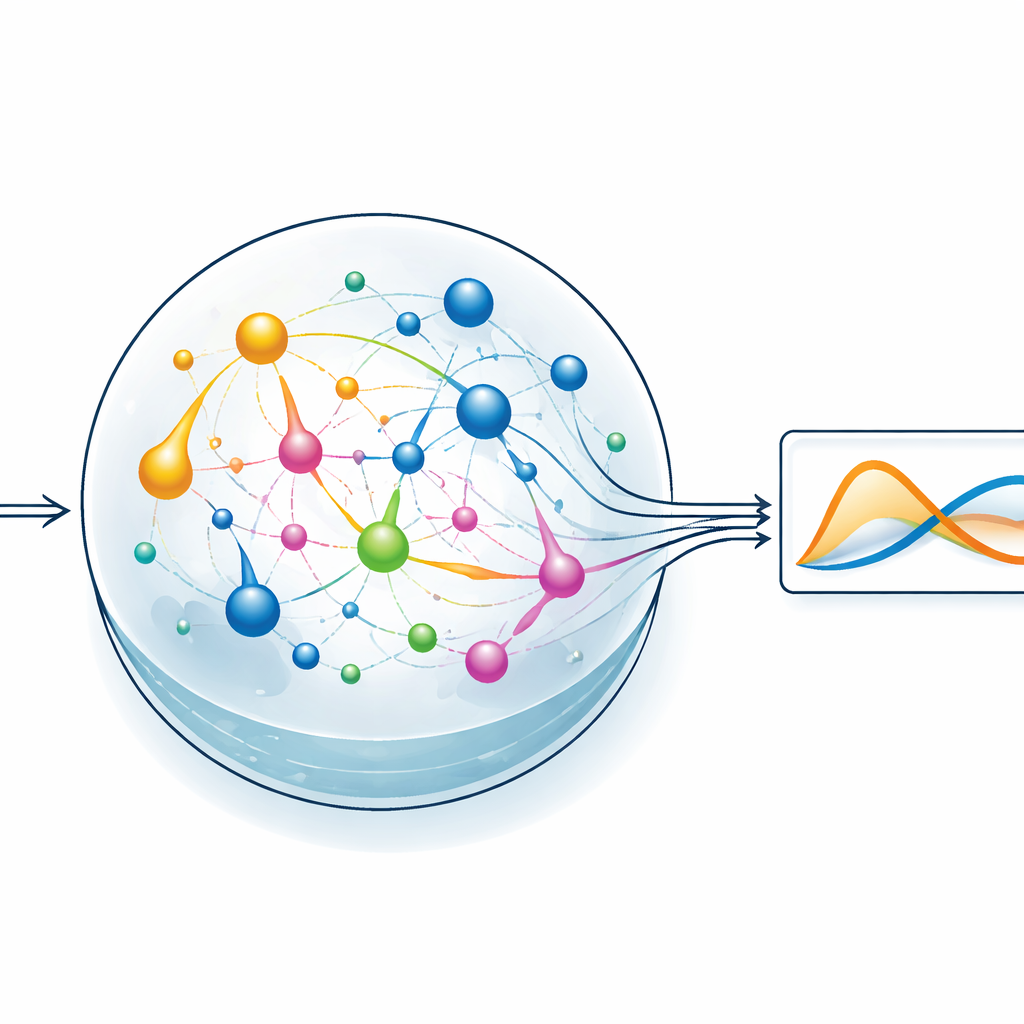
Verso il calcolo a basso consumo energetico in laboratorio
Oltre alle simulazioni, l’articolo guarda avanti ai dispositivi pratici. Discute come la scelta delle reazioni debba bilanciare il consumo energetico, la controllabilità con enzimi o catalizzatori e la capacità di misurare in tempo reale le specie chiave, per esempio con metodi ottici o elettrochimici. Gli autori suggeriscono che future piattaforme microfluidiche potrebbero ospitare reti di reazione accuratamente scelte, con controllo spaziale degli input e sensori integrati. Sebbene rimangano molte sfide ingegneristiche—dalla mappatura delle equazioni alla chimica reale fino alla gestione del rumore e dei limiti di misura—lo studio mostra che sistemi di reazione modesti possono già emulare soluzioni di equazioni differenziali accoppiate. Per un lettore non specialistico, il messaggio centrale è che la chimica stessa può agire come un calcolatore analogico, aprendo la strada a calcoli scientifici che sfruttano i processi a basso consumo energetico che la natura perfeziona da miliardi di anni.
Citazione: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Parole chiave: calcolo chimico, calcolo a basso consumo energetico, reservoir computing, reti di reazione chimica, equazioni differenziali ordinarie