Clear Sky Science · it
Un framework end-to-end per la reattività nella catalisi eterogenea
Perché progettare catalizzatori più rapidamente è importante
La società moderna si basa sui catalizzatori per produrre carburanti, plastiche, fertilizzanti e innumerevoli prodotti di uso quotidiano. Tuttavia trovare catalizzatori migliori è spesso come cercare un ago in un pagliaio, perché ogni materiale può favorire migliaia di possibili reazioni microscopiche contemporaneamente. Questo articolo presenta CARE, un nuovo framework computazionale che usa regole intelligenti e apprendimento automatico per mappare e simulare queste reti di reazioni intricate molto più rapidamente e in modo più completo che in passato. Così facendo, promette di guidare tecnologie energetiche più pulite e processi chimici più efficienti riducendo drasticamente i costi di calcolo.
Sciogliere le vie di reazione affollate
Sulla superficie di un catalizzatore solido, le molecole entranti non seguono semplicemente un unico percorso lineare dal reagente al prodotto. Viaggiano invece attraverso un labirinto di intermedi a vita breve e percorsi concorrenti. I metodi computazionali tradizionali si basano sull’intuito umano per selezionare un numero limitato di possibili step e poi usano calcoli quantistici per valutarne le energie. Questo funziona per reti piccole ma si sgretola rapidamente con l’aumentare della complessità, trascurando percorsi rari che possono governare l’attività a lungo termine, la disattivazione o la selettività. CARE affronta questa sfida costruendo automaticamente reti di reazioni molto vaste a partire da semplici regole costruttive, assicurando che siano incluse tutte le plausibili rotture e formazione di legami tra carbonio, idrogeno e ossigeno, anche quelle che i chimici potrebbero normalmente scartare.
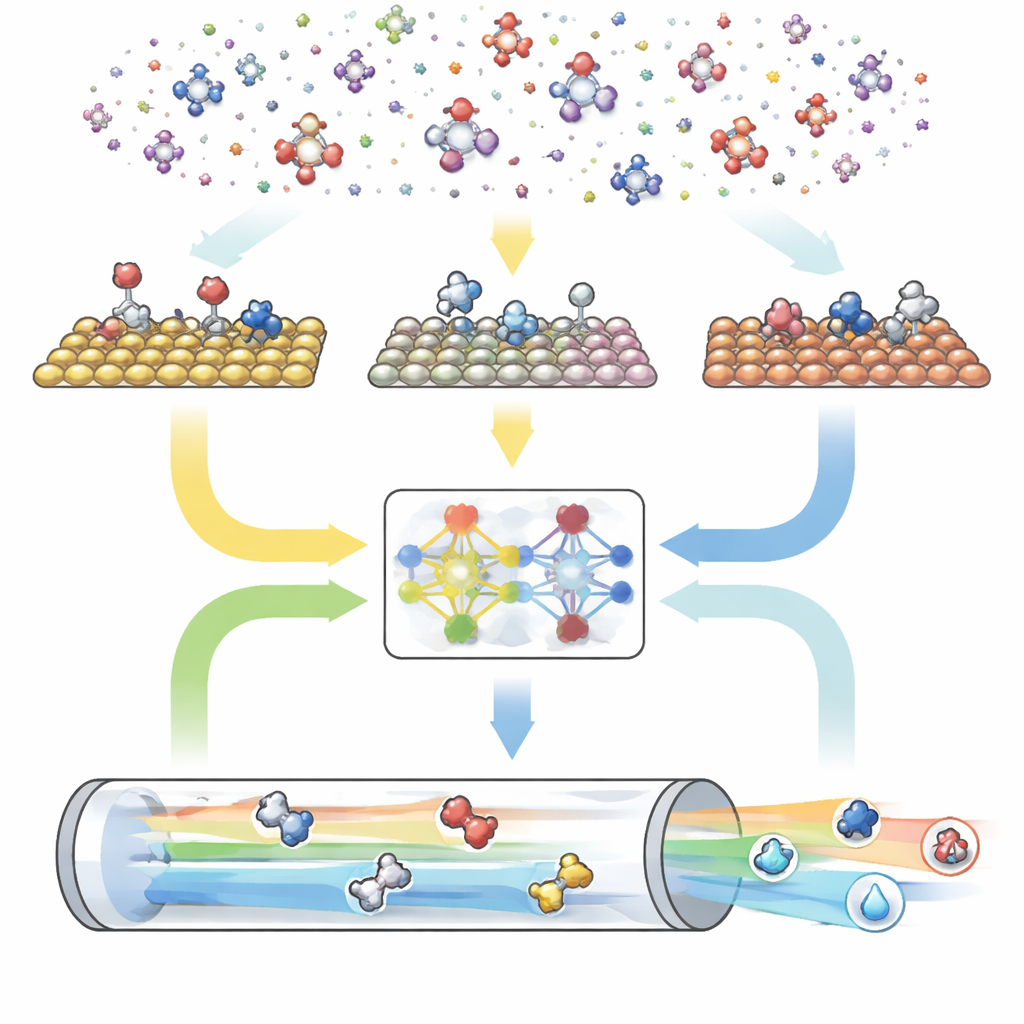
Un motore digitale in tre parti per le reazioni
CARE è strutturato come una pipeline end-to-end con tre moduli principali. Primo, un generatore basato su regole definisce lo “spazio chimico” scegliendo il numero massimo di atomi di carbonio e ossigeno e applicando poi semplici modelli per creare tutte le molecole corrispondenti e le loro forme adsorbite sulla superficie. Secondo, un modulo di valutazione delle energie richiama moderni modelli di machine learning—in particolare una rete grafica neurale chiamata GAME-Net-UQ—per stimare le energie di intermedi e stati di transizione su molte superfici metalliche. Questo modello tratta ogni struttura come una rete di atomi e legami, restituisce sia un’energia sia un’incertezza ed è preciso entro poche decine di decimi di elettronvolt rimanendo leggero e veloce. Terzo, un risolutore microcinetico usa queste energie per calcolare come tutte le reazioni procedono insieme in condizioni realistiche di temperatura, pressione, tensione e pH, prevedendo tassi di reazione complessivi, coperture superficiali e selettività dei prodotti.
Test nel mondo reale: molecole combustibili e chimica climatica
Per dimostrare che CARE non è solo un esercizio teorico, gli autori lo applicano a tre problemi industrialmente rilevanti di difficoltà crescente. Per la decomposizione del metanolo—una reazione importante per lo stoccaggio dell’idrogeno—generano una rete modesta e la valutano su molti catalizzatori metallici e facce cristalline. CARE riproduce la nota tendenza a “volcano” nell’attività e identifica correttamente il rutenio come uno dei migliori performer, in accordo con gli esperimenti, ma impiegando una frazione minima del tempo di calcolo richiesto dai calcoli quantistici completi. Successivamente, passano alla conversione elettrochimica dell’anidride carbonica su rame, concentrandosi su come emergono prodotti a tre atomi di carbonio come il 1-propanolo e il propilene. Includendo passaggi speciali che tengono conto di protoni, elettroni e condizioni di soluzione, CARE cattura come pH e tensione applicata spostino i percorsi e predice correttamente che il 1-propanolo è favorito rispetto al propilene, in eco con studi dettagliati precedenti.
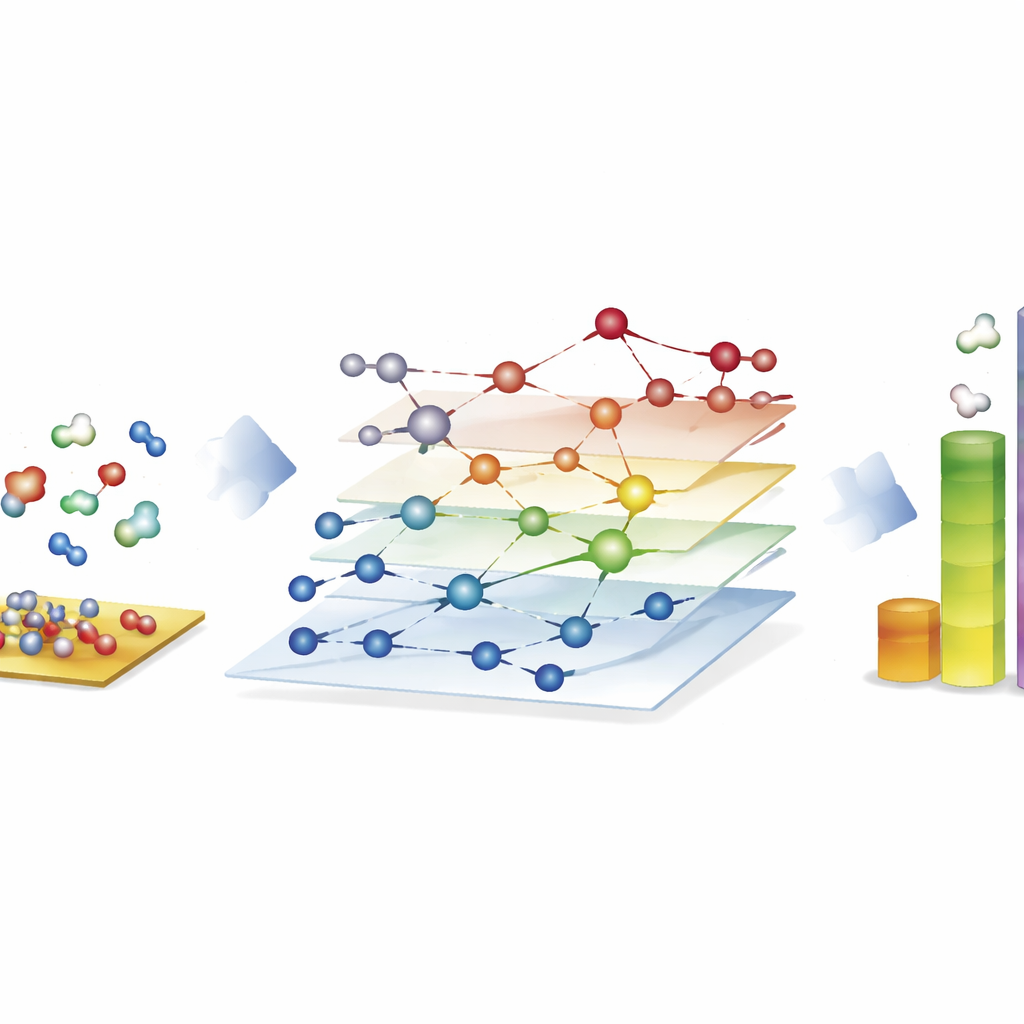
Esplorare enormi reti di reazioni per combustibili sintetici
La dimostrazione più impressionante proviene dal processo di Fischer–Tropsch, che trasforma miscele di monossido di carbonio e idrogeno in idrocarburi a catena lunga per carburanti e prodotti chimici. Qui, gli autori costruiscono reti con quasi 40.000 specie superficiali e circa 370.000 reazioni elementari—ben oltre ciò che gli studi tradizionali basati sulla meccanica quantistica possono esplorare completamente. Usando CARE, valutano tutti gli intermedi e le barriere chiave delle reazioni su superfici di cobalto, ferro, nichel e rutenio in poche ore su hardware standard, con un’accelerazione di circa un milione di volte rispetto ai calcoli quantistici diretti. Le simulazioni microcinetiche su queste reti riproducono tendenze note: cobalto e ferro formano preferenzialmente catene idrocarburiche più lunghe, il ferro produce più diossido di carbonio tramite reazioni laterali e il nichel tende verso una idrogenazione più forte. Sebbene alcuni dettagli, come le rese di metano, rimangano imperfetti, il framework rivela quali passaggi di formazione di legame dominano la crescita delle catene e mette in luce dove i modelli necessitano ancora di perfezionamenti.
Cosa significa questo per i catalizzatori del futuro
Per i non specialisti, il messaggio chiave è che CARE fornisce un modo pratico per esplorare enormi spazi di reazione su superfici catalitiche che prima erano inaccessibili. Automatizzando la generazione delle reti, integrando modelli surrogate di apprendimento automatico rapidi per le energie quantistiche e risolvendo la cinetica risultante in modo efficiente, può classificare i catalizzatori candidati, identificare condizioni operative promettenti e scoprire percorsi inaspettati con molto meno bias umano e spesa computazionale. Pur riconoscendo sfide residue—come una gestione migliore delle superfici affollate, degli effetti del solvente e di reti ancora più vaste—il lavoro indica verso un futuro in cui i computer possono rapidamente selezionare reazioni complesse, dalla riduzione della CO2 al riciclo della plastica e all’upgrading della biomassa, guidando gli esperimenti verso le idee più promettenti anziché lasciare la scoperta al caso e alla prova ed errore.
Citazione: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Parole chiave: catalisi eterogenea, reti di reazioni, apprendimento automatico, modellizzazione microcinetica, sintesi di Fischer–Tropsch