Clear Sky Science · it
Diffusione condizionale con allineamento modale consapevole della località per generare ensemble conformazionali proteici diversificati
Perché il movimento delle proteine è importante
Le proteine nelle nostre cellule non sono sculture rigide; si comportano più come minuscole macchine flessibili che cambiano continuamente forma. Questi cambiamenti di forma possono controllare come gli enzimi catalizzano le reazioni, come i recettori rispondono ai farmaci e come i segnali si propagano nelle cellule. Eppure la maggior parte delle immagini familiari delle proteine mostra una sola “istantanea” strutturale, perdendo il ricco insieme di conformazioni che esistono realmente. Questo articolo presenta Mac-Diff, un metodo di intelligenza artificiale che può generare rapidamente numerose forme realistiche per una data proteina, aiutando gli scienziati a vedere non solo come appare una proteina, ma come respira e si muove.
Dalle singole istantanee agli ensemble in movimento
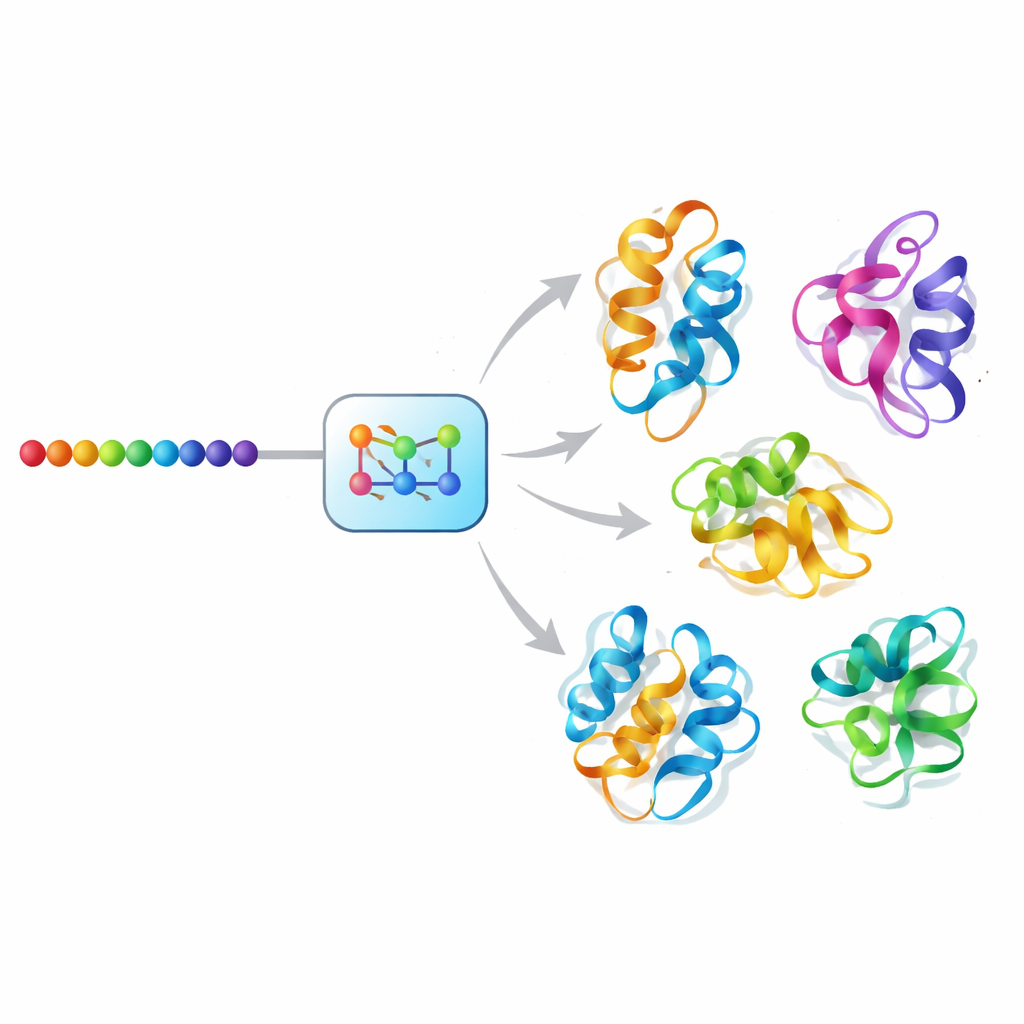
Un approccio di IA “a diffusione” per il movimento delle proteine
Mac-Diff affronta questa sfida utilizzando un modello generativo in stile diffusione, una classe di IA che ha alimentato i recenti progressi nella sintesi di immagini. Invece di denoising di fotografie, Mac-Diff rimuove il rumore da descrizioni geometriche astratte dei backbone proteici. Il modello rappresenta una proteina come una griglia di relazioni a coppie tra i suoi residui — distanze e angoli che sono insensibili a come l’intera molecola è ruotata o traslata. In uno step forward, il sistema aggiunge gradualmente rumore a questi schemi geometrici fino a farli assomigliare a disturbo casuale. Nello step inverso, impara a rimuovere il rumore passo dopo passo, guidato dalla sequenza di amminoacidi della proteina, finché non riemergono geometrie coerenti compatibili con la 3D, che possono poi essere convertite in modelli atomici completi tramite software standard di costruzione strutturale.
Lascare che la sequenza parli alla struttura localmente
Una innovazione chiave risiede nel modo in cui Mac-Diff collega una sequenza lineare di residui ai loro vicini 3D. Lasciare semplicemente che ogni residuo presti attenzione a tutti gli altri residui, come nei modelli text-to-image, sfumerebbe vincoli fisici importanti. Invece, gli autori introducono un meccanismo di attenzione “consapevole della località” che focalizza ogni residuo su un piccolo e probabile vicinato di partner di interazione. Per stimare questi vicinati, Mac-Diff usa tre ingredienti: un modello linguistico per proteine chiamato ESM-2 che codifica il contesto biochimico di ogni residuo; una mappa di contatto che suggerisce quali coppie di residui sono probabili vicine; e una regola semplice che favorisce residui vicini lungo la catena. Questi segnali vengono combinati così che, durante il denoising, il modello utilizzi preferenzialmente informazioni da residui che sono partner fisicamente plausibili, affinando la sua capacità di ricostruire strutture realistiche e flessibili.
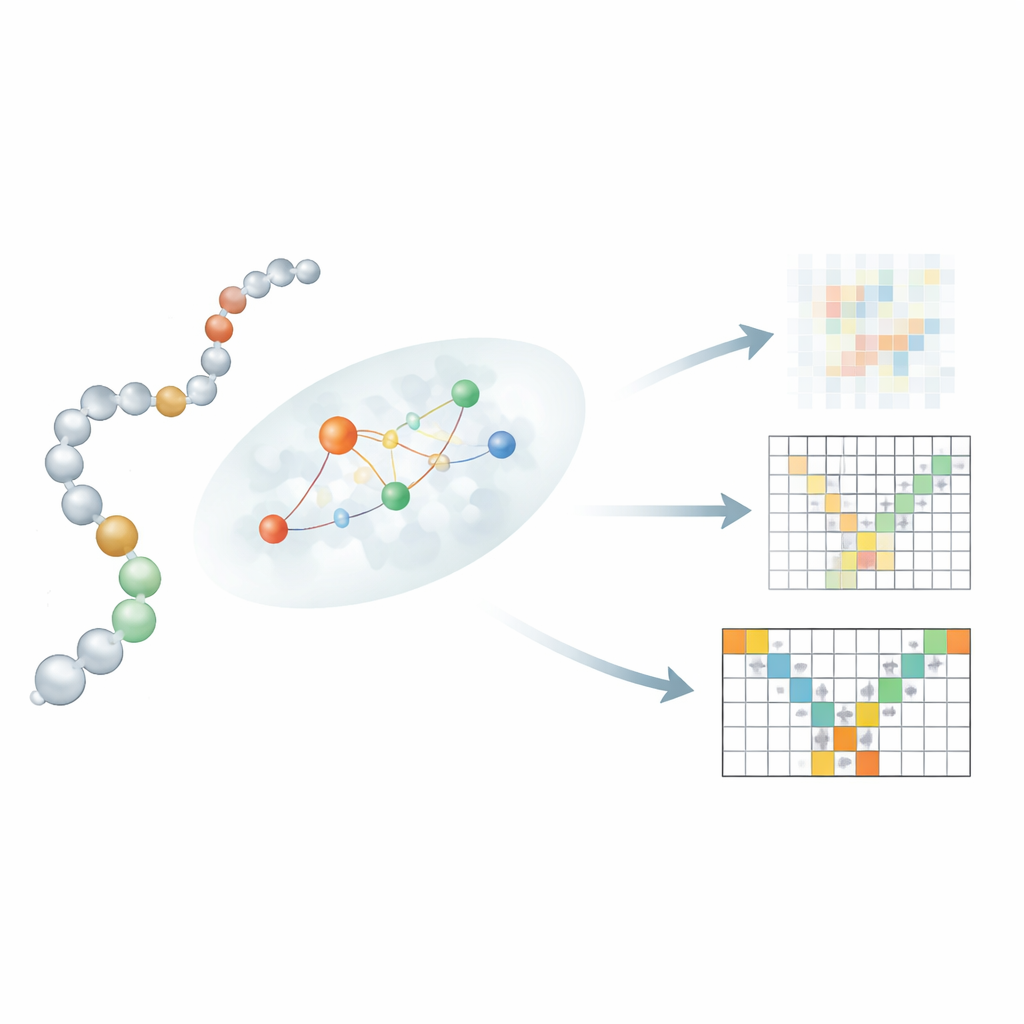
Test contro simulazioni lunghe e proteine che cambiano forma
I ricercatori hanno testato Mac-Diff su due fronti impegnativi. Innanzitutto hanno valutato se fosse in grado di riprodurre l’ampia distribuzione di forme osservata in lunghe e accuratamente calcolate simulazioni di dinamica molecolare di proteine a ripiegamento rapido e di una proteina benchmark classica nota come BPTI. Su diverse misure che confrontano proprietà statistiche degli ensemble generati con i dati di simulazione — come distribuzioni delle distanze all’interno della proteina e compattezza complessiva — Mac-Diff ha eguagliato o superato metodi di IA concorrenti, generando al contempo una più ampia varietà di conformazioni. Ha catturato la maggior parte degli stati “metastabili” chiave identificati nelle simulazioni e ha riprodotto pattern di flessibilità a livello di residuo con alta correlazione, indicando che i suoi ensemble riflettono in modo realistico sia i ripiegamenti globali sia le oscillazioni locali.
Rivelare stati funzionali nascosti
In secondo luogo, il team ha messo alla prova Mac-Diff con proteine note per adottare forme molto diverse durante la loro funzione, inclusi l’enzima adenilato chinasi, che oscilla tra forme aperte e chiuse durante il metabolismo energetico, e un set curato di 40 proteine con due conformazioni determinate sperimentalmente ciascuna. Mac-Diff ha generato soltanto 100 strutture candidate per proteina — molto meno delle tipiche traiettorie di simulazione — eppure ha recuperato la maggior parte degli stati noti con buon accordo geometrico. Nell’adenilato chinasi, ad esempio, ha prodotto sia conformazioni aperte sia chiuse con alta somiglianza alle strutture cristallografiche, mentre diversi metodi popolari tendevano a favorire un solo stato. Il modello ha inoltre funzionato circa mille volte più velocemente delle simulazioni convenzionali su hardware comparabile, rendendo l’esplorazione sistematica della diversità conformazionale molto più pratica.
Cosa significa per la biologia e la medicina
In termini concreti, Mac-Diff trasforma la sequenza di una proteina in una galleria di pose plausibili anziché in un unico ritratto, e lo fa con attenzione a quali parti sono propense a spingere o agganciarsi tra loro in 3D. Campionando questi ensemble con precisione ed efficienza, il metodo offre un modo per sondare come spostamenti sottili di forma sottendano la funzione, per individuare conformazioni rare ma importanti e per cercare tasche di legame per farmaci che emergono solo in stati transitori. Pur non catturando ancora i filmati ordinati nel tempo che forniscono le simulazioni, Mac-Diff porta il paesaggio dinamico delle proteine alla portata di molti più sistemi, promettendo nuove intuizioni nella biologia strutturale, nella progettazione di farmaci e nell’ingegneria proteica.
Citazione: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Parole chiave: dynamics proteiche, modelli di diffusione, ensemble conformazionali, proteine allosteriche, scoperta di farmaci