Clear Sky Science · it
Robustezza senza precedenti dei modelli energetici atomici informati dalla fisica a e oltre la temperatura ambiente
Perché questo è importante per la chimica di tutti i giorni
Le simulazioni al computer sono i cavalli da tiro della chimica moderna e della scienza dei materiali. Consentono agli scienziati di osservare molecole che si torcono, vibrano e collidono in silico invece che in esperimenti costosi e dispendiosi in termini di tempo. Ma quando queste simulazioni si basano sull’apprendimento automatico, possono improvvisamente “esplodere”, producendo forme molecolari impossibili—soprattutto a temperature più elevate. Questo studio introduce un nuovo tipo di modello di apprendimento automatico informato dalla fisica che può eseguire tali simulazioni per tempi estremamente lunghi, a temperature fino a 1000 kelvin, senza collassare.
Da scorciatoie intelligenti a simulazioni fragili
La chimica quantistica tradizionale calcola le energie molecolari con grande accuratezza ma è terribilmente lenta. I campi di forza più semplici sono veloci ma spesso approssimativi. I potenziali appresi mirano a combinare il meglio dei due mondi: apprendono una scorciatoia dalla geometria molecolare all’energia e alle forze, quindi usano quella scorciatoia per guidare la dinamica molecolare. Sulla carta molti di questi modelli sembrano eccellenti, vantando errori medi molto piccoli su set di test standard. In pratica, quei numeri possono essere fuorvianti. Quando le molecole esplorano nuove configurazioni durante una simulazione—soprattutto a temperature elevate—molti modelli vengono spinti oltre l’intervallo di strutture su cui sono stati addestrati. Invece di riportare delicatamente le molecole a forme realistiche, possono predire forze che allungano o schiacciano i legami fino a rendere l’intero sistema non fisico e far abortire la simulazione.
Costruire modelli su blocchi atomici di derivazione quantistica
Gli autori affrontano questa fragilità cambiando ciò che il modello impara e come è guidato da conoscenze fisiche pregresse. Usano un framework chiamato FFLUX, che si basa sull’approccio Interacting Quantum Atoms (IQA). In IQA, una molecola è divisa in “atomi topologici” le cui energie individuali sono determinate direttamente dalla meccanica quantistica. Queste energie atomiche hanno un significato fisico e si sommano all’energia totale della molecola. Invece di apprendere energie di sito arbitrarie, i nuovi modelli a processo gaussiano apprendono queste energie atomiche radicate nella meccanica quantistica, fornendo un’ancora fisica profonda per ogni previsione. Quattro molecole organiche flessibili—glicina e serina con cappe peptidiche, malondialdeide e aspirina—servono da banchi di prova impegnativi per via dei loro numerosi moti interni e della nota difficoltà che pongono ai campi di forza basati su apprendimento automatico esistenti.
Addestrare il modello ad aspettarsi problemi
Una innovazione chiave riguarda come il processo gaussiano è impostato prima ancora di vedere i dati: la sua “funzione di media”, che codifica ciò che il modello presume nelle regioni scarsamente note. La maggior parte dei lavori precedenti imposta questa media a zero, fingendo di fatto che il modello non abbia aspettative pregresse. Gli autori invece spostano deliberatamente questa media verso stati atomici di energia più elevata, mantenendola però fisicamente sensata. Questa scelta progettuale significa che quando il modello è costretto a eseguire extrapolazioni—per esempio quando i legami sono temporaneamente sovraallungati—tende naturalmente a preferire previsioni che penalizzano distorsioni estreme. In test estesi, versioni del modello che differivano solo per questo prior si sono comportate in modo molto diverso. I modelli con medie convenzionali o a bassa energia spesso sopravvivevano meno di un picosecondo prima che una molecola esplodesse o implodesse. Al contrario, la migliore media ad alta energia (chiamata MF5) ha prodotto simulazioni stabili per l’intera finestra di test di un nanosecondo a temperature da 300 a 1000 kelvin per tutte e quattro le molecole.
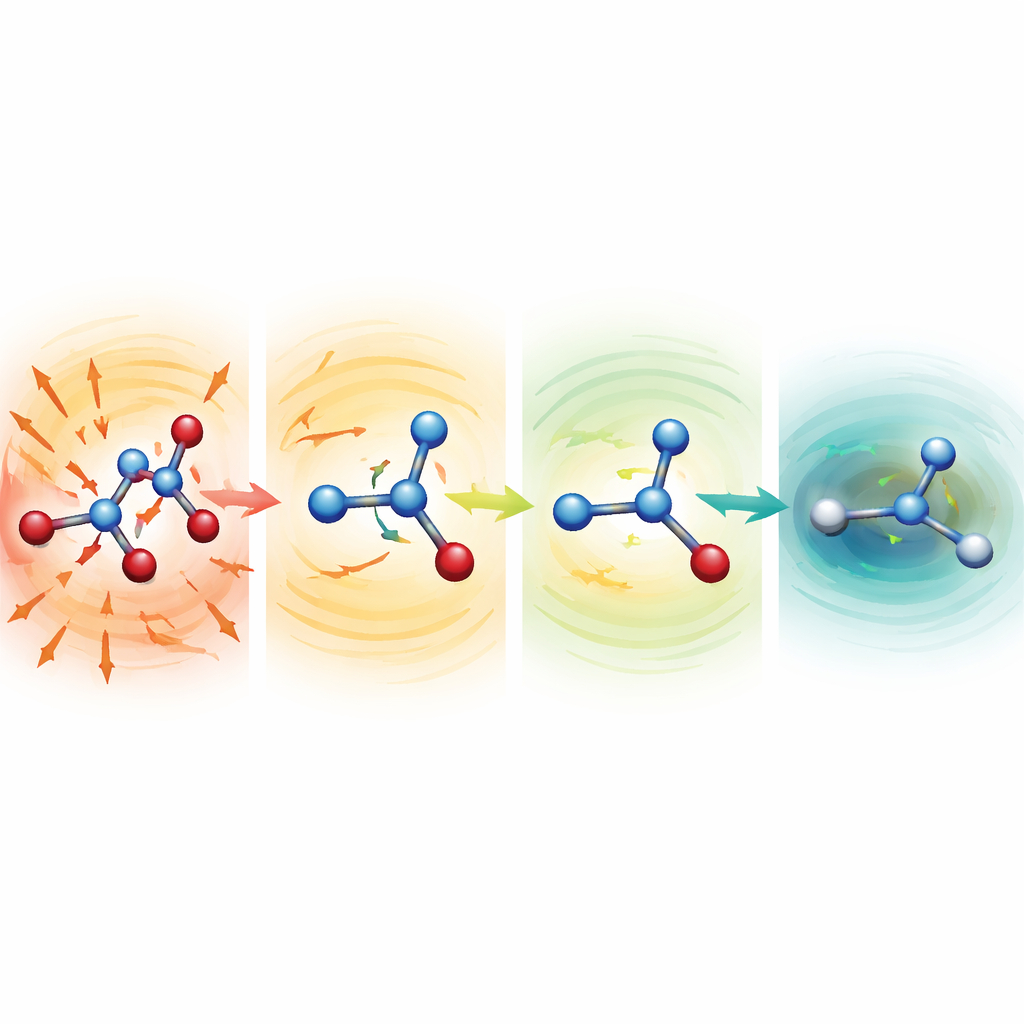
Osservare molecole distorte che si rimediano da sole
Per sondare perché i modelli robusti funzionano così bene, i ricercatori hanno iniziato simulazioni da strutture deliberatamente sfasciate con legami fortemente allungati o compressi. Per serina, aspirina e malondialdeide questi punti di partenza erano a centinaia fino a più di mille kilocalorie per mole sopra una struttura normale—configurazioni che normalmente sarebbero disastrose. Con funzioni di media più deboli, le molecole si disintegravano rapidamente. Con l’impostazione MF5, però, le forze previste puntavano immediatamente in direzioni che accorciavano i legami allungati e allungavano quelli schiacciati. Nel giro di decine o centinaia di passi temporali, le molecole si rilassavano in forme realistiche e poi continuavano a evolvere stabilmente. Il gruppo ha anche dimostrato che, pur non essendo mai stati addestrati sulle forze, gli stessi modelli possono guidare ottimizzazioni di geometria dell’alanina-dipeptide, riproducendo conformazioni a bassa energia note e energie relative entro pochi decimi di kilocaloria per mole, ma a un costo circa 200 volte inferiore rispetto ai calcoli quantistici completi.
Simulazioni lunghe e calde su hardware di uso comune
La robustezza non riguarda soltanto il superare un singolo avvio difficile; riguarda resistere su milioni o miliardi di passi temporali. Gli autori hanno spinto i loro migliori modelli più a fondo eseguendo 50 simulazioni indipendenti a 500 kelvin, ciascuna della durata di 10 nanosecondi, sulle quattro molecole di prova. Nessuna di queste esecuzioni è crashata, fornendo un tempo di simulazione combinato di mezzo microsecondo—insolito per i campi di forza appresi all’avanguardia. Ancora più sorprendente, le simulazioni sono state eseguite in modo efficiente su CPU standard, passo dopo passo rivaleggiando o superando alcuni noti potenziali basati su reti neurali che richiedono GPU potenti. In tutto ciò, le molecole hanno esplorato insiemi ricchi di forme e stati metastabili, mostrando che la robustezza non è stata ottenuta bloccando artificialmente il moto o imponendo strutture rigide.
Cosa significa per il futuro della modellizzazione molecolare
Per i non esperti, il messaggio chiave è che non tutti i modelli di apprendimento automatico a basso errore sono affidabili quando vengono spinti al limite. Radicando i loro modelli in energie atomiche derivate dalla meccanica quantistica e sintonizzando con cura le aspettative intrinseche del modello verso stati ad alta energia, gli autori hanno creato una famiglia di potenziali che producono naturalmente “forze di richiamo”—l’equivalente molecolare di un’imbracatura di sicurezza—che mantengono le simulazioni fisiche anche ad alte temperature e partendo da punti distorti. Questo approccio promette simulazioni più affidabili e più lunghe di molecole complesse e indica possibili estensioni future in cui modelli analoghi informati dalla fisica gestiscono fasi condensate e interazioni sottili come la dispersione, il tutto rimanendo praticabile dal punto di vista computazionale.
Citazione: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Parole chiave: campi di forza apprendimento automatico, robustezza della dinamica molecolare, potenziali a processo gaussiano, modellizzazione informata dalla fisica, energie atomiche quantistiche