Clear Sky Science · it
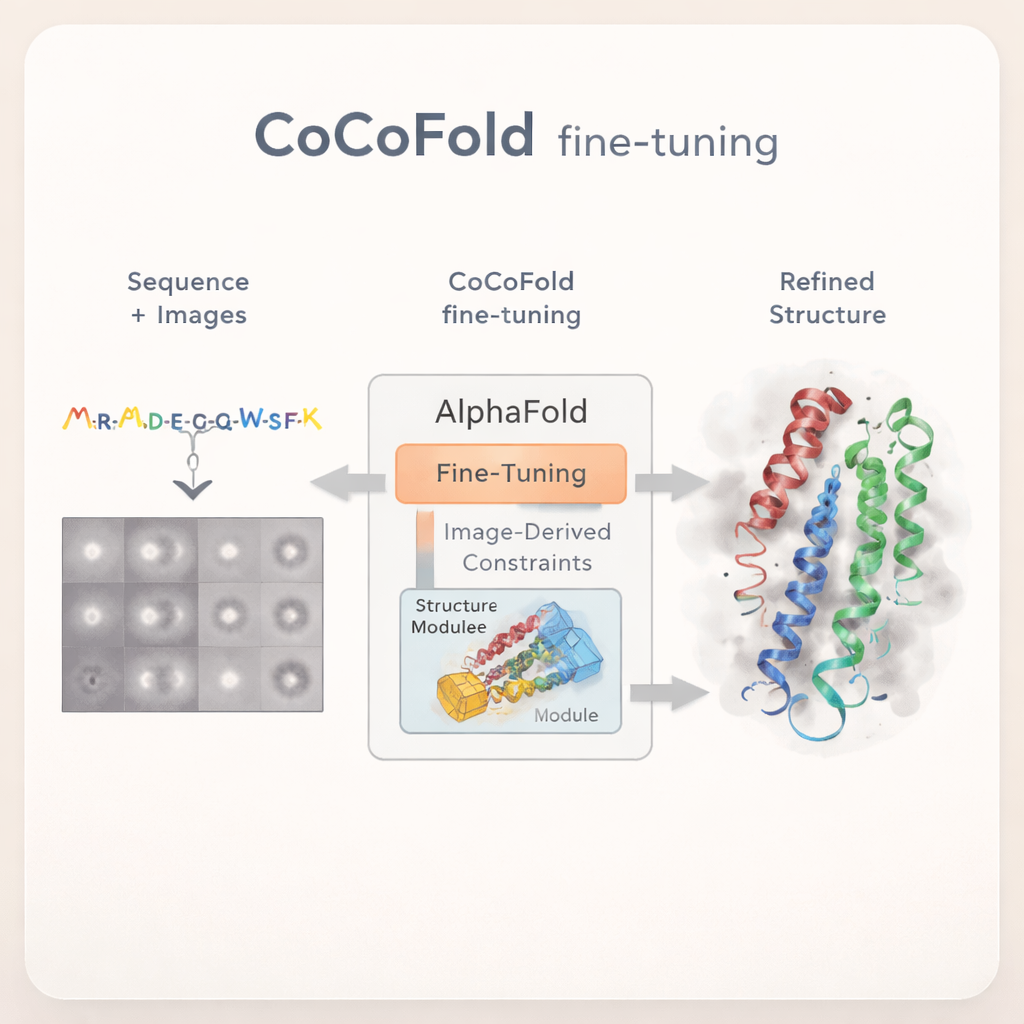
Affinare AlphaFold con osservazioni cryo-EM limitate
Perché le forme delle proteine sono così difficili da vedere
Le proteine sono minuscole macchine molecolari che guidano quasi ogni processo nel nostro corpo, dalla produzione di energia alla trasmissione dei segnali nervosi. Per capire come funzionano — e come i farmaci potrebbero controllarle — gli scienziati hanno bisogno di conoscere con precisione la loro forma tridimensionale. Sono emersi due strumenti potenti per questo compito: la criomicroscopia elettronica (cryo‑EM), che acquisisce numerose immagini sfocate di proteine congelate, e AlphaFold, un sistema di intelligenza artificiale che predice le strutture proteiche a partire dalla sequenza. Tuttavia, in molti esperimenti reali i dati cryo‑EM sono incompleti e le previsioni di AlphaFold non corrispondono sempre alla realtà. Questo articolo presenta CoCoFold, un metodo che insegna ad AlphaFold a “ascoltare” direttamente dati cryo‑EM difficili e a migliorare di conseguenza le sue predizioni.
Quando la macchina fotografica vede troppo poco
La cryo‑EM funziona congelando rapidamente le proteine e acquisendo un numero enorme di particelle individuali da molti angoli, quindi combinando quelle immagini in una mappa 3D. Nella pratica, però, i ricercatori spesso non dispongono di immagini di qualità sufficiente. A volte la proteina compare solo brevemente in uno stato ad alta energia, quindi vengono catturate pochissime particelle. In altri casi le proteine preferiscono certe orientazioni sulla superficie del ghiaccio, lasciando assenti molti angoli di visuale. Entrambi i problemi generano mappe sfocate e incomplete, difficili da tradurre in modelli atomici affidabili. I software esistenti possono adattare le strutture predette da AlphaFold a tali mappe, ma il loro successo dipende fortemente dall’avere dati iniziali nitidi e ad alta risoluzione.
Insegnare ad AlphaFold a imparare dalle immagini grezze
CoCoFold adotta un approccio diverso: invece di fare affidamento su una mappa 3D cryo‑EM completamente ricostruita, utilizza direttamente le immagini 2D delle particelle per affinare AlphaFold. Il metodo parte da una predizione di AlphaFold‑Multimer e mantiene congelata la maggior parte della rete originale, preservandone la conoscenza generale della piegatura proteica. Solo la parte finale deputata alla costruzione della struttura è autorizzata a cambiare. Viene aggiunto un “adapter” leggero per immettere informazioni derivate dalle immagini cryo‑EM in questo modulo strutturale, spingendo delicatamente il modello verso conformazioni compatibili con i dati sperimentali evitando deviazioni eccessive dalla fisica nota delle proteine.
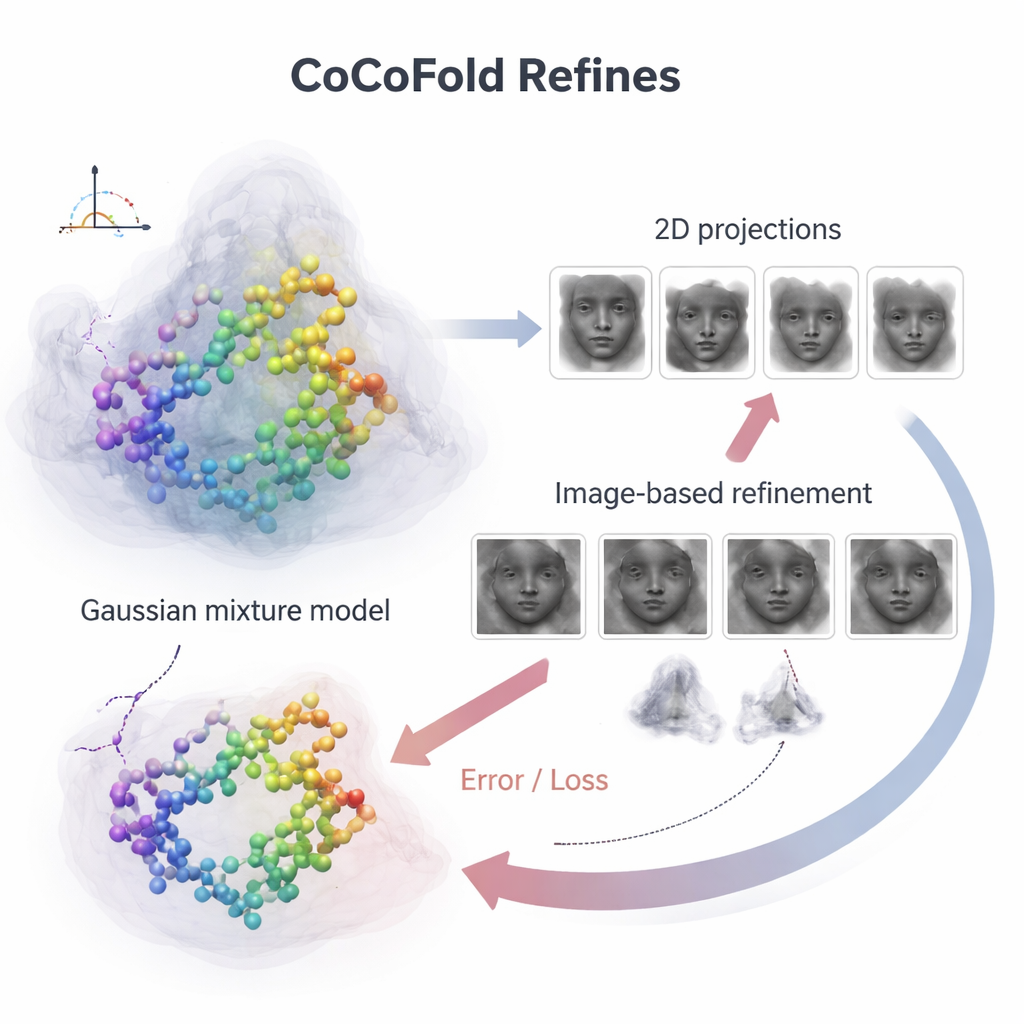
Trasformare le immagini in feedback strutturale
Per connettere i singoli atomi proteici alle rumorose immagini del microscopio, CoCoFold costruisce un’immagine morbida e flessibile della struttura prevista usando blob tridimensionali sovrapposti, noti come una miscela gaussiana. Da questa rappresentazione simula come la proteina apparirebbe al microscopio nelle stesse direzioni di visuale e condizioni di imaging dell’esperimento reale. Questi scatti simulati vengono quindi confrontati con le particelle cryo‑EM effettive, anello per anello nel dominio delle frequenze, per valutare quanto corrispondano. Qualsiasi disallineamento diventa un segnale di feedback che fluisce attraverso la rete, aggiustando leggermente sia il modello proteico sia la rappresentazione della densità. Dopo l’addestramento, il modello atomico viene ulteriormente ripulito con un passo di raffinamento basato sulla fisica per rimuovere conflitti geometrici locali.
Mantenere accuratezza quando i dati scarseggiano o sono distorti
Gli autori hanno testato CoCoFold su diversi dataset sperimentali e simulati progettati per imitare i due problemi principali della cryo‑EM: troppe poche particelle e grandi lacune negli angoli di visuale. In queste condizioni difficili, gli strumenti standard — incluse altre metodologie di deep learning che dipendono da mappe ricostruite — tendevano a perdere regioni della proteina, spostare eliche o perdere dettagli fini man mano che le mappe diventavano più sfocate. CoCoFold, al contrario, ha prodotto costantemente modelli che corrispondevano più fedelmente e più completamente alle strutture di riferimento note. I suoi errori restavano contenuti anche quando il numero di particelle veniva drasticamente ridotto o quando mancavano ampi coni di direzioni di visuale, suggerendo che l’apprendimento diretto dalle immagini grezze preserva informazioni cruciali che gli approcci basati sulla mappa scartano.
Cosa significa per la biologia strutturale futura
Per i non specialisti, il messaggio chiave è che CoCoFold funge da traduttore tra potenti predizioni AI e dati sperimentali imperfetti. Invece di fidarsi esclusivamente di AlphaFold o della cryo‑EM, permette ai due approcci di informarsi a vicenda, specialmente nei casi difficili in cui gli esperimenti offrono solo una visione parziale. Nei casi semplici con dati abbondanti e di alta qualità, gli strumenti guidati dalla mappa funzionano ancora molto bene. Ma quando le particelle sono rare o mancano orientazioni — situazioni comuni quando si inseguono stati proteici fugaci o fragili — CoCoFold offre un modo per recuperare modelli atomici affidabili da informazioni che altrimenti andrebbero perse.
Citazione: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Parole chiave: cryo-EM, AlphaFold, struttura proteica, deep learning, biologia strutturale