Clear Sky Science · it
Insufficienza energetica mitocondriale alla base della neuropatia sensoriale correlata a FLVCR1
Quando i nervi del dolore rimangono senza energia
Alcune persone nascono quasi incapaci di percepire il dolore. A prima vista può sembrare una benedizione, ma si trasforma presto in una condanna: privi del dolore come segnale di allarme, accumulano ustioni, fratture, infezioni e persino cecità. Questo studio esamina una rara forma di tali disturbi della perdita del dolore e individua un colpevole sorprendente: piccoli impianti energetici all’interno delle cellule nervose la cui produzione di energia va gravemente fuori rotta.
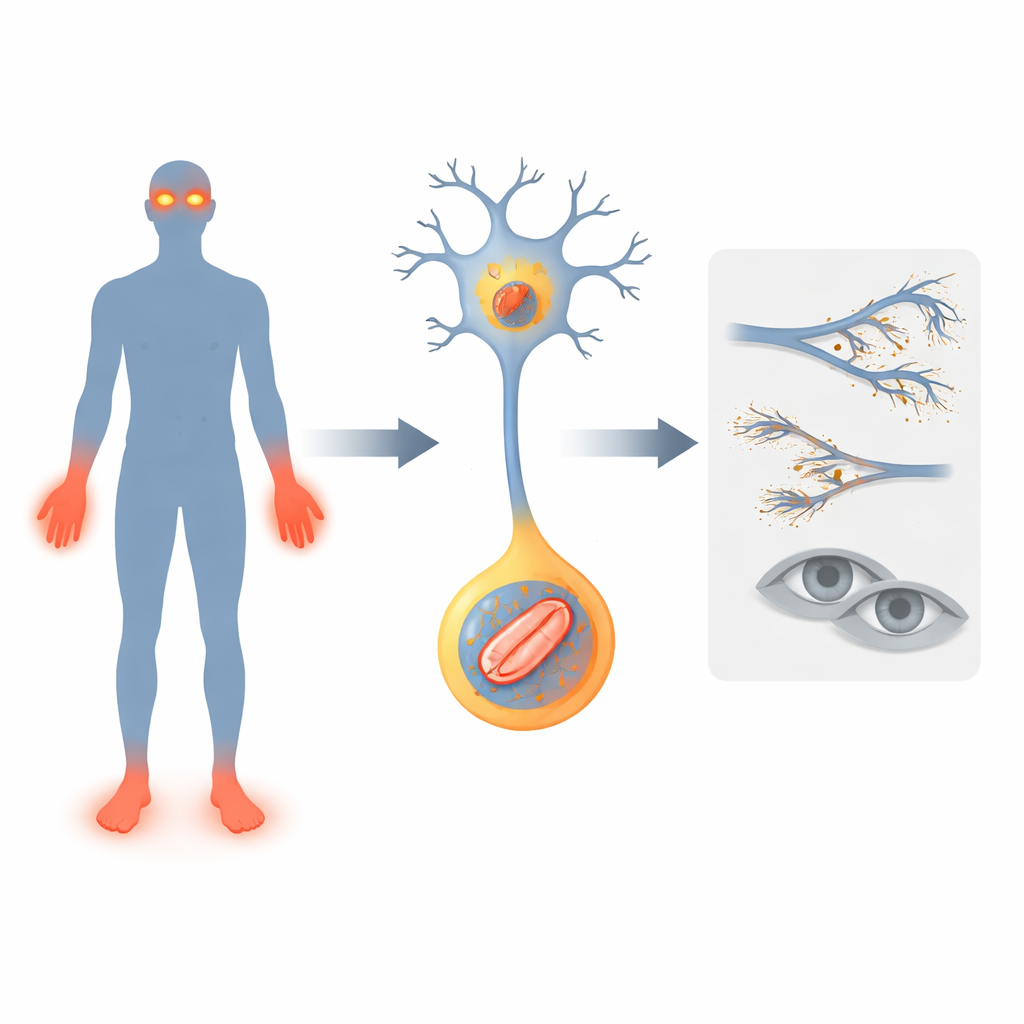
Un gene che spegne le campane d’allarme
I ricercatori si concentrano su un gene chiamato FLVCR1, già associato a rari disturbi nervosi in cui le persone perdono la sensibilità al dolore, sviluppano un’andatura instabile e talvolta una perdita visiva progressiva. Descrivono due nuovi pazienti con varianti in FLVCR1. Entrambi i bambini presentarono problemi precoci: ritardo nelle tappe motorie, cadute frequenti, infezioni profonde e mutilazione di dita delle mani e dei piedi perché le lesioni non venivano avvertite. Uno sviluppò anche una malattia degenerativa dell’occhio chiamata retinite pigmentosa, con conseguente cecità notturna. Questi casi amplianno il quadro delle manifestazioni legate a difetti di FLVCR1 nell’uomo e rafforzano l’idea che questo gene sia vitale per mantenere in vita i nervi sensoriali e le cellule fotosensibili della retina.
Modellare la malattia in piccoli pesci
Per esplorare come FLVCR1 influenzi i nervi sensoriali in sviluppo, il team si è rivolto al zebrafish, i cui embrioni trasparenti permettono di osservare direttamente le cellule nervose. Hanno ridotto i livelli della versione ittica del gene, flvcr1a, usando strumenti genetici. I pesci con flvcr1a diminuito avevano minori gangli delle radici dorsali, ammassi di neuroni che rilevano il tatto e il dolore lungo la colonna vertebrale. A livello di comportamento, questi pesci si muovevano meno spontaneamente e nuotavano solo brevi distanze quando la coda veniva toccata delicatamente, suggerendo una risposta sensoriale attenuata. Poiché modelli murini precedenti morivano troppo presto per analizzare i loro nervi sensoriali, questi zebrafish forniscono il primo sistema vivo in cui i difetti nervosi e comportamentali correlati a FLVCR1 possono essere seguiti in dettaglio.
Tre vie compromesse convergono sugli organelli energetici
FLVCR1 è localizzato nelle membrane cellulari e gestisce diverse sostanze chiave. Lavori precedenti suggerivano ruoli nell’elaborazione della colina (un componente delle membrane lipidiche), dell’eme (il pigmento contenente ferro che alimenta molti enzimi) e nel flusso di calcio tra compartimenti cellulari. Gli scienziati hanno raccolto cellule della pelle (fibroblasti) da quattro pazienti portatori di diverse mutazioni di FLVCR1 e le hanno confrontate con cellule di persone sane e portatori asintomatici. Hanno riscontrato che le cellule dei pazienti avevano livelli di colina più bassi e membrane cellulari più fluide, cambiamenti che potrebbero disturbare il delicato ambiente lipidico richiesto dai mitocondri, gli organelli produttrici di energia. Hanno anche scoperto che un enzima cruciale per la sintesi dell’eme all’interno dei mitocondri, ALAS1, era meno attivo, sebbene il contenuto totale di eme apparisse quasi normale. Contemporaneamente, i siti di contatto fisico tra reticolo endoplasmatico e mitocondri—dove il calcio normalmente fluisce nei mitocondri—erano più corti e meno frequenti, e l’ingresso di calcio nei mitocondri risultava ridotto. Tre problemi—carenza di colina, produzione rallentata di eme e trasferimento di calcio indebolito—indicavano tutti verso una prestazione mitocondriale compromessa.
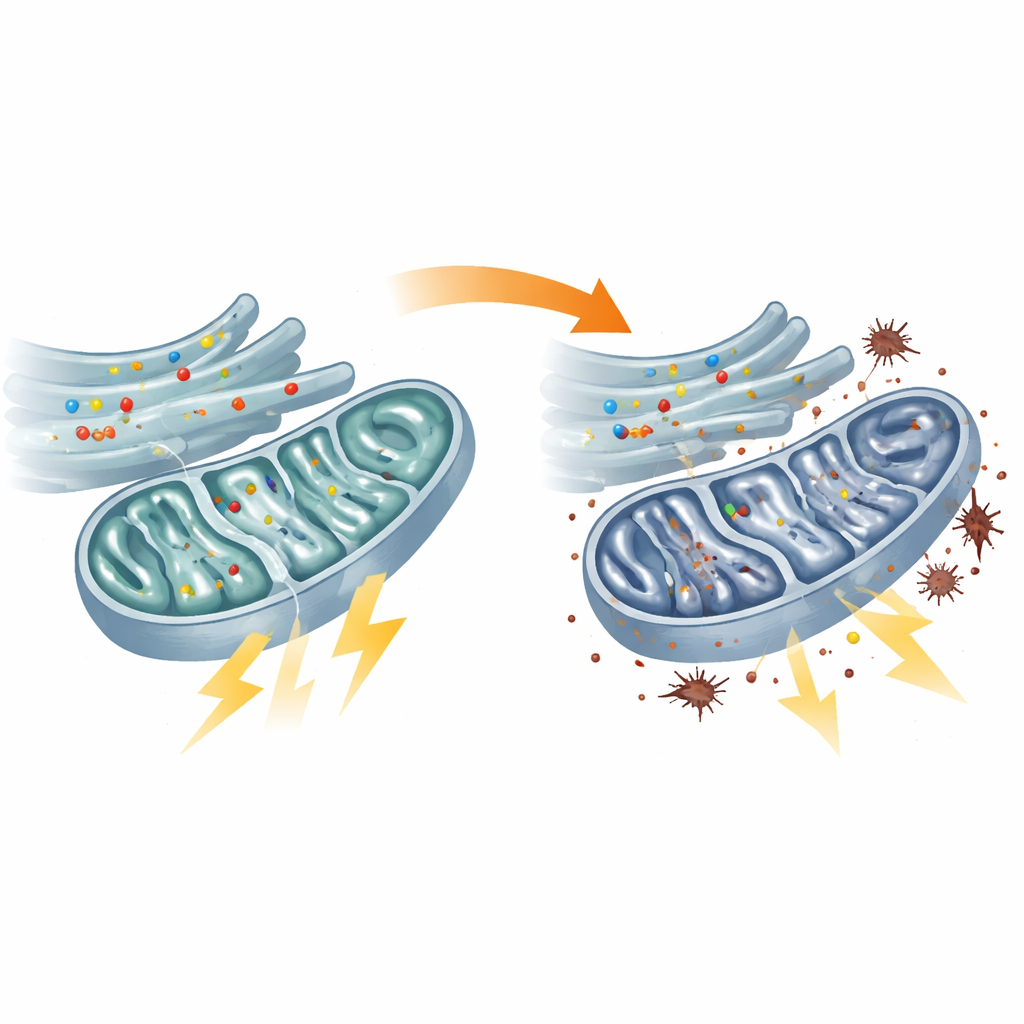
Mitocondri affamati e sistemi di riserva sovraccarichi
Test diretti del metabolismo energetico confermarono che i mitocondri nei fibroblasti dei pazienti erano sottoperformanti. Il centro di elaborazione del carburante noto come ciclo TCA funzionava più lentamente, diversi dei suoi enzimi chiave risultavano meno attivi e la catena di reazioni che normalmente converte il carburante in ATP, la valuta energetica della cellula, era attenuata. Di conseguenza, i livelli di ATP all’interno dei mitocondri calarono. Le cellule cercarono di compensare aumentando la glicolisi, una via meno efficiente di combustione degli zuccheri al di fuori dei mitocondri. Questo cambiamento di strategia energetica ebbe un costo: elettroni fuoriusciti dalla macchina mitocondriale stressata innescarono livelli più elevati di perossidazione lipidica, una forma di danno ossidativo alle membrane cellulari. Difetti simili furono osservati nei zebrafish con flvcr1a ridotto, collegando direttamente il malfunzionamento mitocondriale al modello animale della neuropatia sensoriale.
Indizi per futuri trattamenti potenziando l’energia cellulare
In modo incoraggiante, alcuni di questi difetti possono essere attenuati in laboratorio. Quando il team aumentò artificialmente l’ingresso di calcio nei mitocondri sovraesprimendo una proteina canale chiamata MCU nelle cellule dei pazienti, la produzione di energia migliorò e i segni di danno ossidativo diminuirono. Fornire alle cellule un precursore dell’eme, l’acido 5-aminolevulinico (ALA), migliorò inoltre l’attività del ciclo TCA, la funzione della catena respiratoria e i livelli di ATP, sebbene un’esposizione prolungata ad ALA sia risultata dannosa in studi precedenti. L’aggiunta di colina normalizzò la fluidità delle membrane e contribuì a ridurre il danno lipidico, ma apportò solo guadagni modesti a breve termine nella produzione energetica. Questi esperimenti di recupero suggeriscono che nessuna singola via è esclusivamente responsabile; piuttosto, una rete di alterazioni nella gestione di colina, eme e calcio spinge i mitocondri in una performance cronica ridotta.
Perché queste scoperte sono importanti per i pazienti
Tracciando le conseguenze delle mutazioni di FLVCR1 dalle molecole alle cellule fino all’organismo intero, questo lavoro propone che l’insufficienza energetica mitocondriale sia una forza motrice dietro questa forma di neuropatia da perdita del dolore e i suoi problemi visivi associati. I nervi sensoriali e i fotorecettori sono particolarmente esigenti dal punto di vista energetico perché mantengono assoni lunghi o rinnovano continuamente le strutture fotosensibili, rendendoli particolarmente vulnerabili quando la resa mitocondriale viene meno. Il modello di zebrafish e le cellule derivate dai pazienti offrono ora terreni di prova pratici per terapie che rafforzino il metabolismo mitocondriale. Sebbene trattamenti come l’integrazione di colina, il potenziamento controllato dell’eme o farmaci che aumentano l’apporto di calcio mitocondriale richiederanno una valutazione accurata in modelli animali e studi clinici, il messaggio centrale è chiaro: ripristinare l’alimentazione energetica dei neuroni fragili potrebbe un giorno aiutare a proteggere chi nasce senza il più importante segnale d’allarme naturale: il dolore.
Citazione: Bertino, F., Zanin Venturini, D.I., Grasso, E. et al. Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy. Commun Biol 9, 429 (2026). https://doi.org/10.1038/s42003-026-09691-y
Parole chiave: neuropatia sensoriale, disfunzione mitocondriale, FLVCR1, insensibilità al dolore, metabolismo energetico nervoso