Clear Sky Science · it
Guida pratica alle tecnologie di sequenziamento RNA single-cell mirato
Perché osservare le singole cellule è importante
Ogni cellula del tuo corpo contiene lo stesso DNA, eppure cellule diverse si comportano in modi molto differenti. Lo fanno attivando o disattivando specifici geni e modificando le molecole di RNA in modi sottili. Il sequenziamento RNA single-cell moderno può leggere quali RNA sono presenti in migliaia di cellule contemporaneamente, ma attualmente perde gran parte del messaggio. Questa recensione spiega dove le tecniche attuali perdono informazioni e come nuovi metodi “mirati” vengono sviluppati per concentrarsi sulle parti più importanti delle molecole di RNA a scopo di ricerca, diagnosi e progettazione terapeutica.
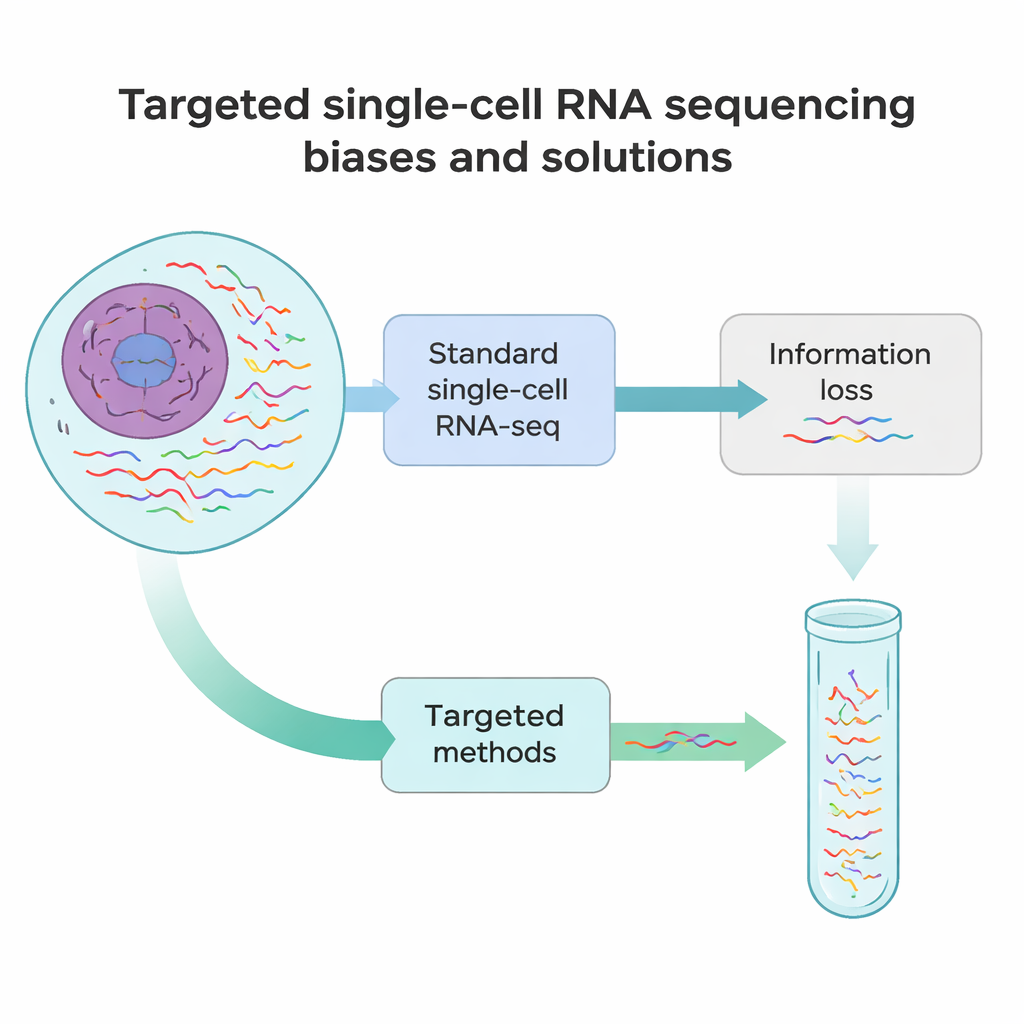
Dove i metodi odierni sono carenti
Il sequenziamento RNA single-cell standard funziona un po’ come scattare un’istantanea rapida di ogni messaggio in una cellula anziché girare un filmato completo. Nella maggior parte degli esperimenti viene rilevato solo circa il 10–40% di tutti gli RNA di una cellula, e si legge soltanto l’inizio o la fine. Ciò significa che molti RNA rari ma importanti — come i marcatori che definiscono l’identità cellulare o le versioni geniche portatrici di mutazioni responsabili di malattie — vengono facilmente persi. Inoltre, diversi passaggi tecnici, dallo scindere i tessuti in singole cellule al copiare l’RNA in DNA e amplificarlo, introducono bias sistematici. Alcuni RNA vengono troncati precocemente, altri risultano sovrarappresentati e altri scompaiono completamente dai dati.
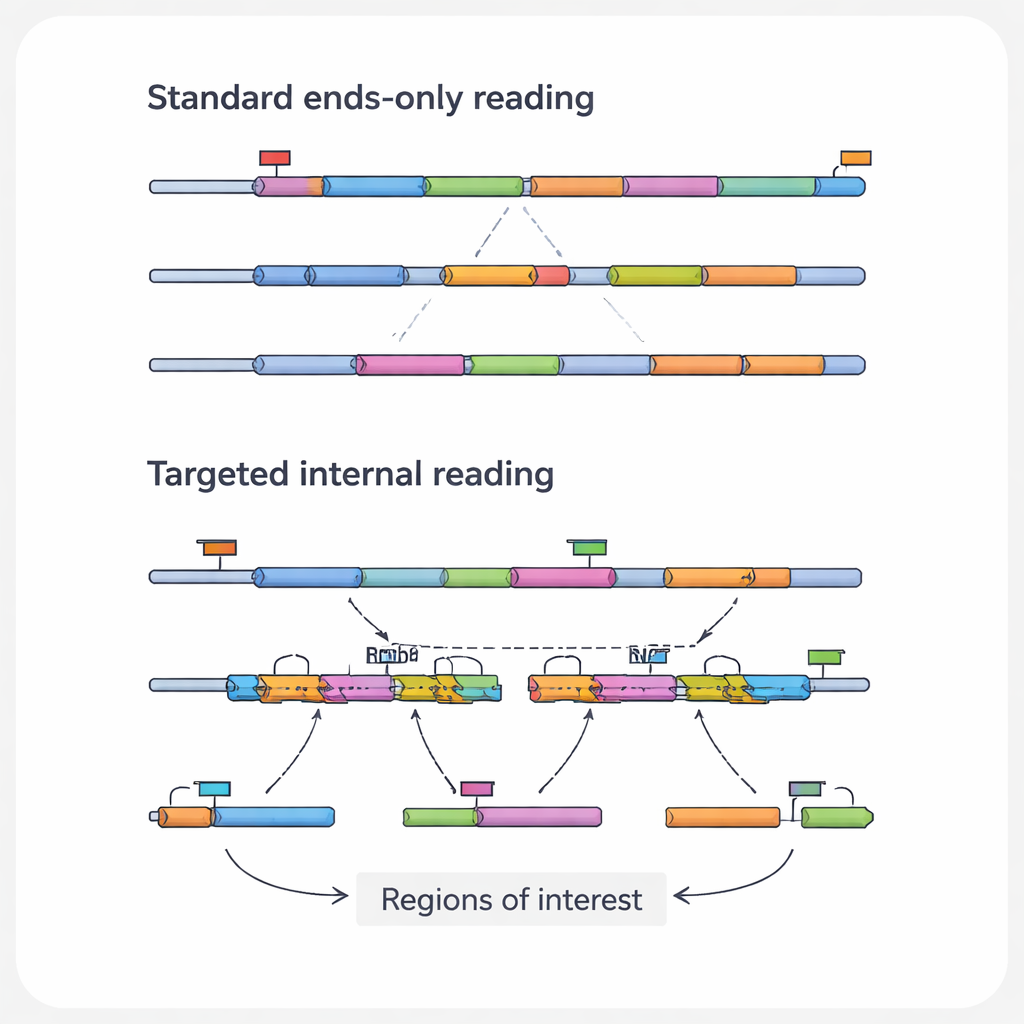
Perché i dettagli interni dell’RNA contano
Le informazioni di maggior rilevanza clinica in una molecola di RNA spesso risiedono nelle sue regioni interne, non alle estremità che i metodi standard leggono. Queste sezioni interne possono contenere mutazioni puntiformi che guidano il cancro, punti di fusione in cui due geni si sono uniti anormalmente, o giunzioni di splicing che generano diverse varianti proteiche dallo stesso gene. Possono inoltre conservare le impronte di strumenti di editing genico come CRISPR. Gli autori chiamano queste caratteristiche specifiche “regioni di interesse” e gli RNA che le portano “trascritti di interesse”. Poiché le piattaforme ad alto rendimento comuni leggono principalmente le punte degli RNA, tendono a trascurare regolarmente questi dettagli cruciali, specialmente nei trascritti lunghi o a bassa abbondanza.
Nuovi modi per puntare il riflettore
Per superare questi punti ciechi, i ricercatori hanno sviluppato una famiglia di approcci di sequenziamento RNA single-cell mirato. Invece di cercare di leggere ogni RNA allo stesso modo, questi metodi arricchiscono deliberatamente trascritti o regioni selezionate. Alcune strategie riprogettano le sfere di cattura in modo che si leghino a sequenze interne dell’RNA anziché soltanto alla coda, estraendo i messaggi scelti nella libreria fin dal primo passaggio. Altre aggiungono primer personalizzati che iniziano la sintesi in un punto interno, oppure passaggi PCR extra che amplificano specificamente una lista ristretta di geni da una libreria esistente. Un ulteriore gruppo utilizza sonde di DNA che ibridano RNA target o loro copie e poi li catturano, spesso con semplici etichette chimiche. Ogni categoria comporta compromessi tra sensibilità, numero di cellule, numero di target e costo, ma tutte hanno lo stesso obiettivo: recuperare più dettagli significativi dagli stessi o da meno read di sequenziamento.
Applicazioni dai virus ai tumori
Questi metodi mirati stanno già rimodellando diverse aree della biologia e della medicina. Nelle infezioni possono finalmente catturare RNA virali o batterici privi della coda poli(A) che i protocolli standard richiedono, rivelando quali cellule ospiti li contengono e come alterano l’attività genica dell’ospite. Nel cancro, il sequenziamento single-cell mirato può individuare quali tipi cellulari portano specifiche mutazioni o geni di fusione e collegarli a programmi genici alterati, aiutando a spiegare perché alcune cellule diventano resistenti alle terapie. Altri metodi si concentrano sullo splicing alternativo, scoprendo quali tipi cellulari utilizzano quali isoforme, o su popolazioni cellulari rare e marker sottili che altrimenti resterebbero sotto la soglia di rilevamento. In schermate CRISPR in pool, una migliore cattura delle guide RNA permette ai ricercatori di associare ogni perturbazione genetica alla precisa risposta cellulare.
Scegliere lo strumento giusto e cosa verrà dopo
Poiché ora esiste un corredo affollato di approcci mirati, gli autori propongono un albero decisionale per aiutare i ricercatori a scegliere un metodo. Le domande chiave includono se è necessario il profilo dell’intero trascrittoma, quante regioni o geni devono essere presi di mira, quanto distano queste regioni dalle estremità dell’RNA e quante cellule si possono processare. Guardando al futuro, sostengono che i maggiori guadagni deriveranno dal migliorare i passaggi di cattura iniziali, dall’ampliare le strategie ingegnose basate su sonde e dal combinare il targeting con le piattaforme emergenti di letture lunghe e di sequenziamento diretto dell’RNA. Finché non sarà pratico leggere ogni RNA in ogni cellula da un estremo all’altro, il sequenziamento RNA single-cell mirato rimarrà essenziale per vedere le parti del messaggio cellulare che contano di più per la biologia e la malattia.
Citazione: Moro, G., Brunner, E. & Basler, K. A practical guide to targeted single-cell RNA sequencing technologies. Commun Biol 9, 250 (2026). https://doi.org/10.1038/s42003-026-09675-y
Parole chiave: sequenziamento RNA single-cell, sequenziamento mirato, trascrittomica, mutazioni del cancro, trascrittomica spaziale