Clear Sky Science · it
La demetilasi degli istoni KDM7A regola negativamente la polarizzazione dei macrofagi fibrotici e la progressione della fibrosi polmonare
Perché i polmoni cicatrizzati riguardano tutti
Quando nei polmoni si formano cicatrici persistenti, respirare diventa una difficoltà quotidiana. Questa condizione, nota come fibrosi polmonare, colpisce milioni di persone e attualmente non ha una cura—solo farmaci che rallentano il danno. In questo studio, i ricercatori hanno scoperto un «freno» molecolare finora nascosto all’interno delle cellule immunitarie chiamate macrofagi che aiuta a mantenere sotto controllo la formazione di cicatrici polmonari. Comprendere questo freno potrebbe aprire la strada a nuovi trattamenti non solo per la fibrosi polmonare, ma potenzialmente anche per altre malattie in cui cicatrizzazione dannosa e infiammazione incontrollata vanno di pari passo.
Il racconto di cellule immunitarie camaleontiche
I macrofagi sono cellule immunitarie di prima linea che pattugliano i tessuti, rimuovono detriti e favoriscono la riparazione dei danni. Ma sono anche camaleontiche: in alcune situazioni diventano combattenti pro-infiammatori, mentre in altre si trasformano in riparatori che possono favorire la formazione di cicatrici. Un tipo specifico che promuove la cicatrizzazione, chiamato macrofagi profibrotici (Fib-Mac), è fortemente associato alla fibrosi polmonare. Queste cellule producono molecole che attivano i fibroblasti, i quali depositano poi collageno e altri componenti della matrice in eccesso, irrigidendo progressivamente il polmone. Gli autori volevano capire come le «impostazioni» genetiche all’interno dei macrofagi decidano se questi assumano l’identità pericolosa di Fib-Mac o restino in stati più equilibrati e protettivi.

Un freno epigenetico nascosto nel genoma
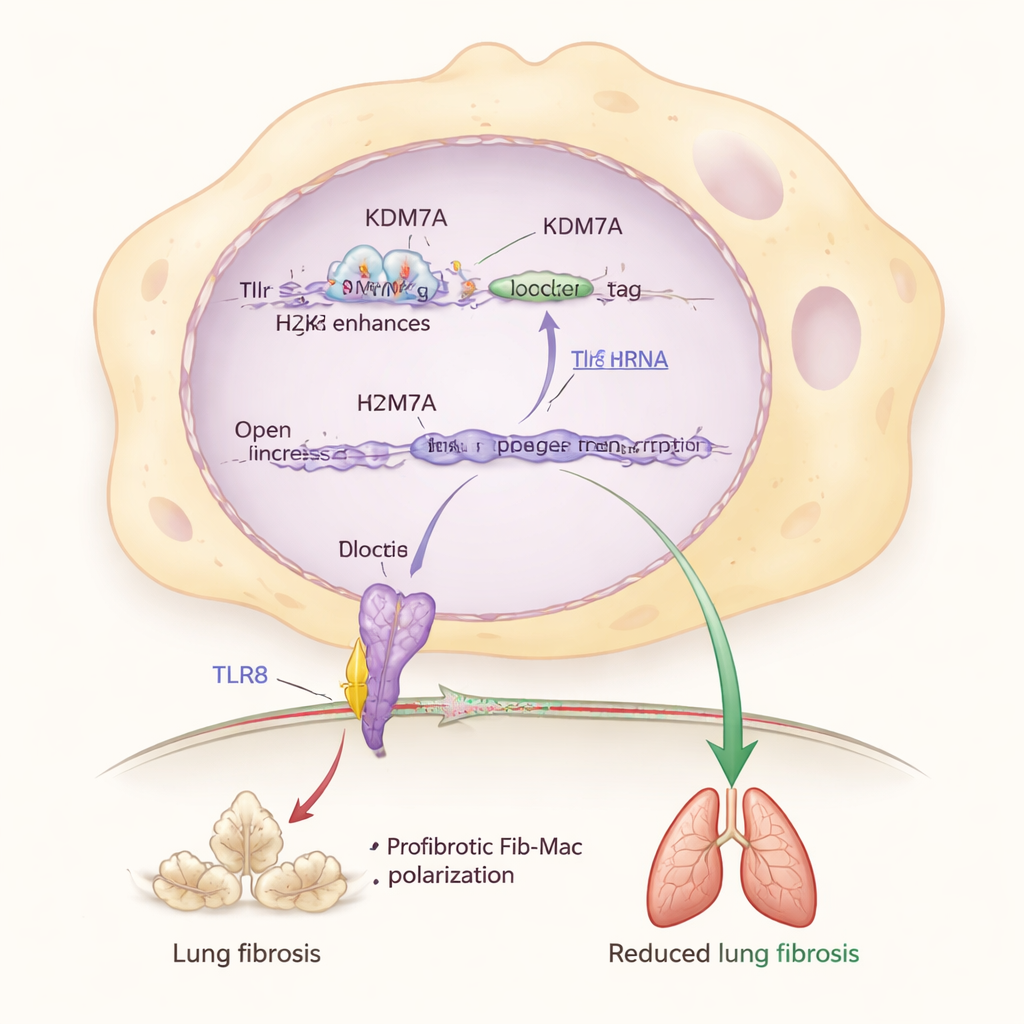
Il gruppo ha iniziato analizzando centinaia di regolatori epigenetici noti—proteine che modulano quanto strettamente il DNA è impacchettato e quali geni sono attivi o spenti. Utilizzando il sequenziamento dell’RNA in macrofagi umani e murini, hanno scoperto che un enzima chiamato KDM7A veniva fortemente attivato quando i macrofagi venivano spinti verso uno stato fibrotico e di riparazione. KDM7A è una «demetilasi degli istoni»: rimuove specifici segni chimici dalle proteine istoniche attorno alle quali il DNA è avvolto. Questo schema suggeriva che KDM7A potesse agire come un freno di retroazione integrato, attivato proprio quando i macrofagi cominciano a deviare verso un’identità che promuove le cicatrici.
Per verificare questa ipotesi, i ricercatori hanno usato topi privi del gene Kdm7a e hanno indotto un danno polmonare con il farmaco chemioterapico bleomicina, un modello standard di fibrosi polmonare. Nelle fasi iniziali dopo il danno, il tessuto polmonare appariva simile nei topi normali e in quelli privi di Kdm7a. Ma dopo tre settimane, i topi senza Kdm7a mostravano cicatrici molto più estese, collasso degli alveoli e punteggi Ashcroft più elevati che quantificano la fibrosi. I geni coinvolti nella produzione di collagene e in altre vie correlate alla fibrosi risultavano più attivi in questi animali knock-out, confermando che la perdita di Kdm7a rende i polmoni più vulnerabili a una formazione di cicatrici prolungata e dannosa.
Come KDM7A indirizza i macrofagi lontano da un destino che favorisce le cicatrici
Con il sequenziamento dell’RNA a singola cellula, gli autori si sono concentrati sulle singole cellule polmonari provenienti da topi danneggiati. Hanno scoperto che in assenza di Kdm7a, un particolare sottogruppo di macrofagi nel tessuto di supporto del polmone si espandeva in maniera significativa e acquisiva una forte firma Fib-Mac, esprimendo geni come Arg1, Spp1 e Trem2. Esperimenti aggiuntivi su macrofagi coltivati hanno mostrato che la rimozione di Kdm7a aumentava i geni marker dei Fib-Mac e riorientava il metabolismo cellulare verso vie che favoriscono la produzione di collagene e l’attivazione sostenuta. In altre parole, KDM7A normalmente limita sia i programmi genetici sia quelli metabolici che spingono i macrofagi verso uno stato promotore di fibrosi.
Approfondendo, i ricercatori hanno identificato un partner chiave in questo sistema frenante: una proteina sensore chiamata TLR8, che rileva frammenti di RNA all’interno delle cellule immunitarie. Hanno osservato che KDM7A aiuta a mantenere il gene Tlr8 attivo rimuovendo un marchio chimico repressivo (H3K27me2) da una regione potenziatrice vicina a Tlr8. Quando Kdm7a veniva disattivato, questo marchio si accumulava, i livelli di Tlr8 calavano e le caratteristiche Fib-Mac si intensificavano. Ridurre direttamente Tlr8 nei macrofagi li spingeva anch’esso verso un’identità fibrotica, mentre attivare o sovraesprimere TLR8 li riportava indietro, anche in assenza di Kdm7a. Questo colloca la via KDM7A–TLR8 al centro di un circuito molecolare che protegge i polmoni da una cicatrizzazione eccessiva.
Dai polmoni invecchiati alla malattia umana
Per collegare questi risultati agli esseri umani, il gruppo ha esaminato tessuto polmonare di pazienti con malattia polmonare fibrotica. Rispetto al tessuto di controllo non malato, i polmoni fibrotici contenevano molti più macrofagi con marcatori Fib-Mac, ma quelle stesse cellule mostravano livelli marcatamente ridotti di KDM7A e TLR8. La rianalisi di dataset a singola cellula esistenti di pazienti con fibrosi polmonare idiopatica ha confermato questo schema: all’aumentare della firma Fib-Mac, l’espressione di KDM7A diminuiva. I ricercatori hanno anche esplorato un grande atlante murino e riscontrato che l’espressione di Kdm7a e Tlr8 nei macrofagi declinava con l’età nei maschi, riflettendo il maggior rischio di fibrosi polmonare negli uomini anziani. Ciò suggerisce che l’indebolimento del freno KDM7A–TLR8 legato all’età e al sesso potrebbe contribuire a spiegare chi è più vulnerabile a cicatrici polmonari severe.
Cosa significa per i trattamenti futuri
In termini semplici, questo lavoro mostra che il nostro sistema immunitario porta con sé un meccanismo di sicurezza interno che impedisce alle cellule riparatrici di diventare troppo zelanti e trasformarsi in promotori di cicatrici permanenti. KDM7A, agendo tramite TLR8, impedisce ai macrofagi di bloccarsi in una modalità profibrotica e aiuta così a mantenere un tessuto polmonare flessibile e funzionale dopo un danno. Quando questo sistema viene meno—per perdita genetica, invecchiamento o altri fattori—i macrofagi sono più inclini a diventare «amplificatori di cicatrici», peggiorando la fibrosi. Rivelando questo freno epigenetico, lo studio indica nuove strategie terapeutiche: farmaci che potenzino l’attività di KDM7A, ne mimino gli effetti o stimolino con cura TLR8 potrebbero un giorno integrare le terapie antifibrotiche esistenti e offrire una migliore protezione contro la progressiva e limitante fibrosi polmonare.
Citazione: Funagura, N., Koga, T., Etoh, K. et al. Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression. Commun Biol 9, 309 (2026). https://doi.org/10.1038/s42003-026-09610-1
Parole chiave: fibrosi polmonare, macrofagi, epigenetica, KDM7A, TLR8