Clear Sky Science · it
haCCA: integrazione multi-modulo di trascrittomi e metabolomi spaziali a spot
Perché mappare le molecole in situ conta
I nostri corpi sono costituiti da innumerevoli piccoli quartieri cellulari, ciascuno con la propria combinazione di geni attivi e sostanze chimiche. Fino a poco tempo fa, gli scienziati dovevano studiare queste molecole dopo aver polverizzato i tessuti in una pasta uniforme, perdendo ogni nozione di “dove” fossero. Questo articolo presenta un nuovo metodo computazionale, chiamato haCCA, che unisce due potenti tecniche di imaging per permettere ai ricercatori di vedere in situ come geni e piccole molecole sono distribuiti nei tessuti e nei tumori reali. Un tale tipo di mappa può rivelare pattern patologici nascosti e suggerire trattamenti più mirati.
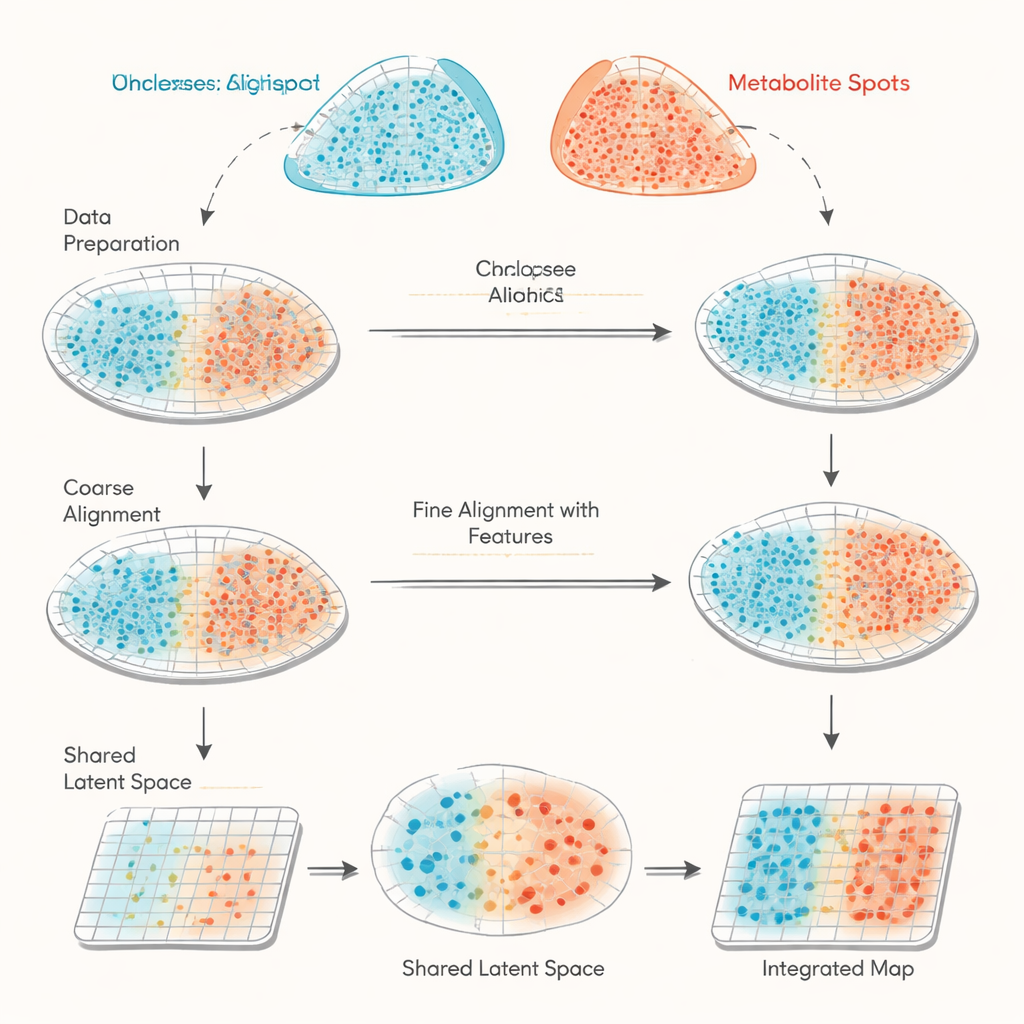
Due diverse viste dello stesso tessuto
Lo studio si concentra sulla combinazione di dati provenienti da due metodi spaziali sempre più usati in biologia. La trascrittomica spaziale registra quali geni sono attivi in migliaia di piccoli spot distribuiti su una sezione di tessuto. L’imaging di spettrometria di massa MALDI misura le quantità di molte piccole molecole, come metaboliti e lipidi, su reticoli di spot altrettanto densi. Il problema è che questi due strumenti non misurano esattamente le stesse posizioni né lo stesso insieme di caratteristiche, perciò i loro dati assomigliano a due mappe disallineate con legende diverse. Gli approcci esistenti cercano per lo più di allineare la forma delle sezioni tissutali basandosi solo sulle coordinate, cosa che può essere imprecisa e priva di un modo per verificare quanto l’allineamento sia effettivamente riuscito.
Un modo più intelligente per mettere in linea le mappe molecolari
haCCA (abbreviazione di hierarchical anchor-guided canonical correlation analysis) affronta questa sfida combinando la geometria con la biologia. Prima esegue un’«allineamento morfologico» in due fasi delle griglie di spot delle due tecnologie. Esperti umani selezionano alcuni punti di riferimento corrispondenti nelle immagini del tessuto per correggere grossolanamente spostamenti e rotazioni, quindi una fase automatica rifinisce gli outlier vicino a bordi strappati o a pezzi mancanti. Successivamente, il metodo individua coppie di “ancore” di spot che sono vicine nello spazio e si trovano in regioni localmente uniformi, rendendole probabili rappresentanti della stessa area tessutale. A partire da questi spot ancora, haCCA calcola quali geni e metaboliti tendono a variare insieme e li sintetizza in una rappresentazione condivisa a bassa dimensione che cattura i loro pattern congiunti più forti.
Trasformare le correlazioni in un’immagine tessutale unificata
Con in mano sia le coordinate spaziali sia la rappresentazione molecolare condivisa, haCCA risolve un problema di ottimizzazione per decidere quanto sia probabile che ciascuno spot genico sia abbinato a ciascuno spot metabolico. Questa fase è progettata per mantenere gli spot vicini nello spazio ma anche simili nel loro profilo combinato gene–metabolita. Il risultato finale è un “piano di trasporto” che collega ogni punto di un dataset al miglior partner nell’altro, producendo una mappa multi-modale integrata. Su dati di test costruiti accuratamente—dove le relazioni vere sono note—gli autori mostrano che ogni fase del flusso di lavoro (allineamento grossolano, raffinato e abbinamento sensibile alle caratteristiche) migliora progressivamente tre misure indipendenti di accuratezza. Rispetto ad altri strumenti che si basano principalmente sulla geometria, haCCA ottiene costantemente un allineamento migliore e un trasferimento più fedele delle etichette di regione.
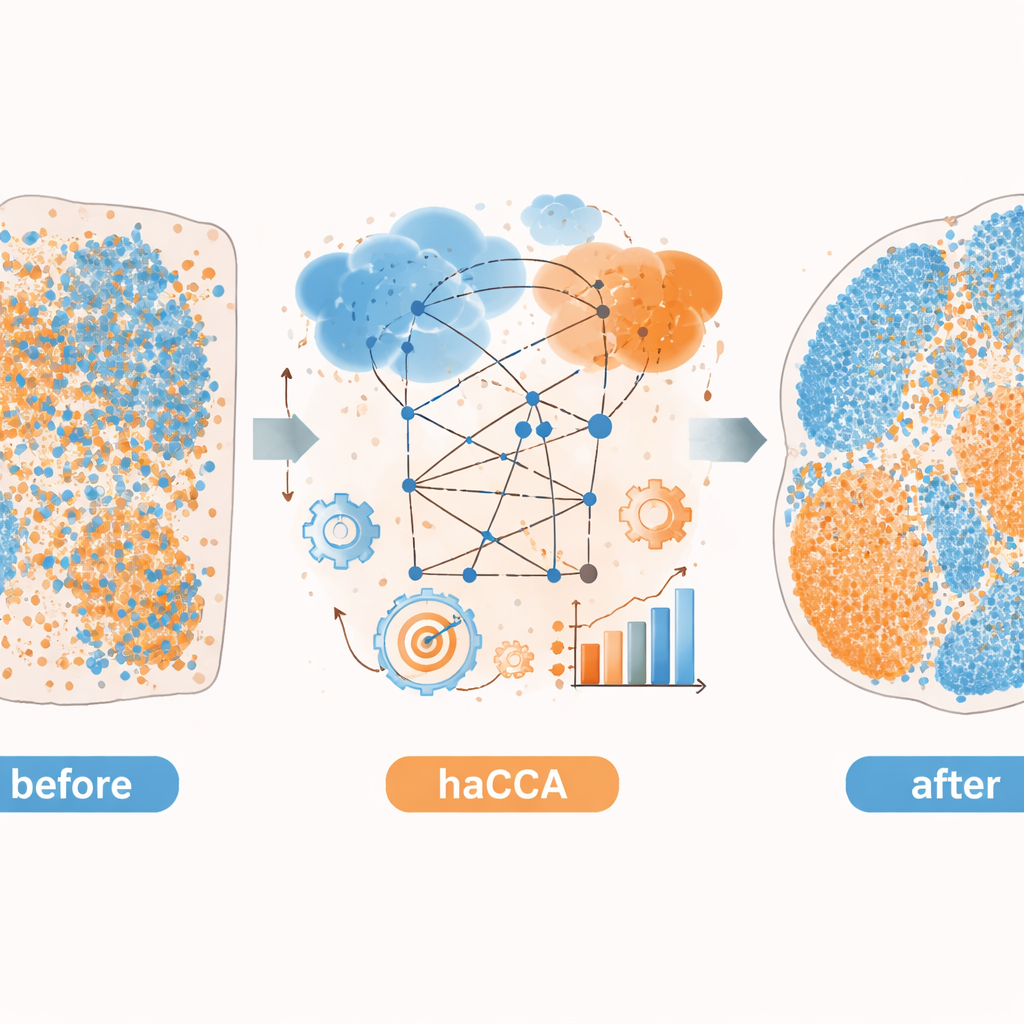
Rivelare biologia nascosta nei tumori cerebrali e del fegato
Gli autori applicano poi haCCA a tessuti reali di cervello di topo e tumori epatici. Per il cervello, integrano dati commerciali di trascrittomica spaziale con immagini di metaboliti ottenute nella stessa sezione o in sezioni adiacenti. Il metodo preserva territori metabolici noti e ricostruisce sovrapposizioni attese, come la co-localizzazione della dopamina con il gene che codifica il suo enzima chiave. Clusterizzando insieme geni e metaboliti, scoprono che i dati combinati separano sottoregioni tessutali più sfumate rispetto a ciascuna modalità da sola. In un modello preclinico di colangiocarcinoma intraepatico, un tipo di tumore del fegato, utilizzano haCCA per confrontare tumori che possono o non possono formare trappole extracellulari per neutrofili—strutture a rete rilasciate dalle cellule immunitarie. Le mappe integrate rivelano che, quando queste trappole sono presenti, un gene chiamato Scd1 e gli acidi grassi a esso associati sono arricchiti nelle regioni maligne, suggerendo uno spostamento verso un metabolismo lipidico alterato nel tumore.
Cosa significa per la ricerca futura
In termini pratici, haCCA è come allineare foto aeree scattate con fotocamere diverse—una sensibile ai contorni degli edifici, l’altra alle firme termiche—per ottenere un’immagine più nitida di ciò che accade in ogni isolato. Mettendo insieme con precisione dove i geni sono attivi e dove si accumulano i metaboliti chiave, questo flusso di lavoro aiuta gli scienziati a profilare entrambe le facce del comportamento cellulare contemporaneamente: le istruzioni e la chimica risultante. L’approccio migliora i metodi di allineamento precedenti, è confezionato in uno strumento Python accessibile e può essere esteso ad altre tecnologie spaziali. Man mano che tali mappe integrate diventano più comuni, potrebbero approfondire la nostra comprensione di come tumori e altri tessuti organizzano il loro metabolismo, rispondono ai trattamenti e si evolvono nel tempo.
Citazione: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Parole chiave: multi-omica spaziale, trascrittomica, metabolomica, metabolismo tumorale, integrazione dei dati