Clear Sky Science · it
Determinazione ab initio delle stabilità di fase nei solidi dinamicamente disordinati: disordine rotazionale C2 in Li2C2
Perché questo solido che si sposta è importante
Molte tecnologie moderne si basano su solidi in grado di modificare silenziosamente la loro struttura interna quando vengono riscaldati o compressi. Questi cambiamenti, detti transizioni di fase, sono centrali in idee come il raffreddamento a stato solido e batterie più sicure. Questo studio esamina un composto semplice, il carburo di litio (Li2C2), che passa da una forma ordinata a una forma più irrequieta e dinamicamente disordinata al crescere della temperatura. Seguendo questa trasformazione atomo per atomo in simulazioni al computer, gli autori mostrano come la «irrequietezza» interna di piccole unità molecolari possa ribaltare l’equilibrio tra due strutture cristalline.
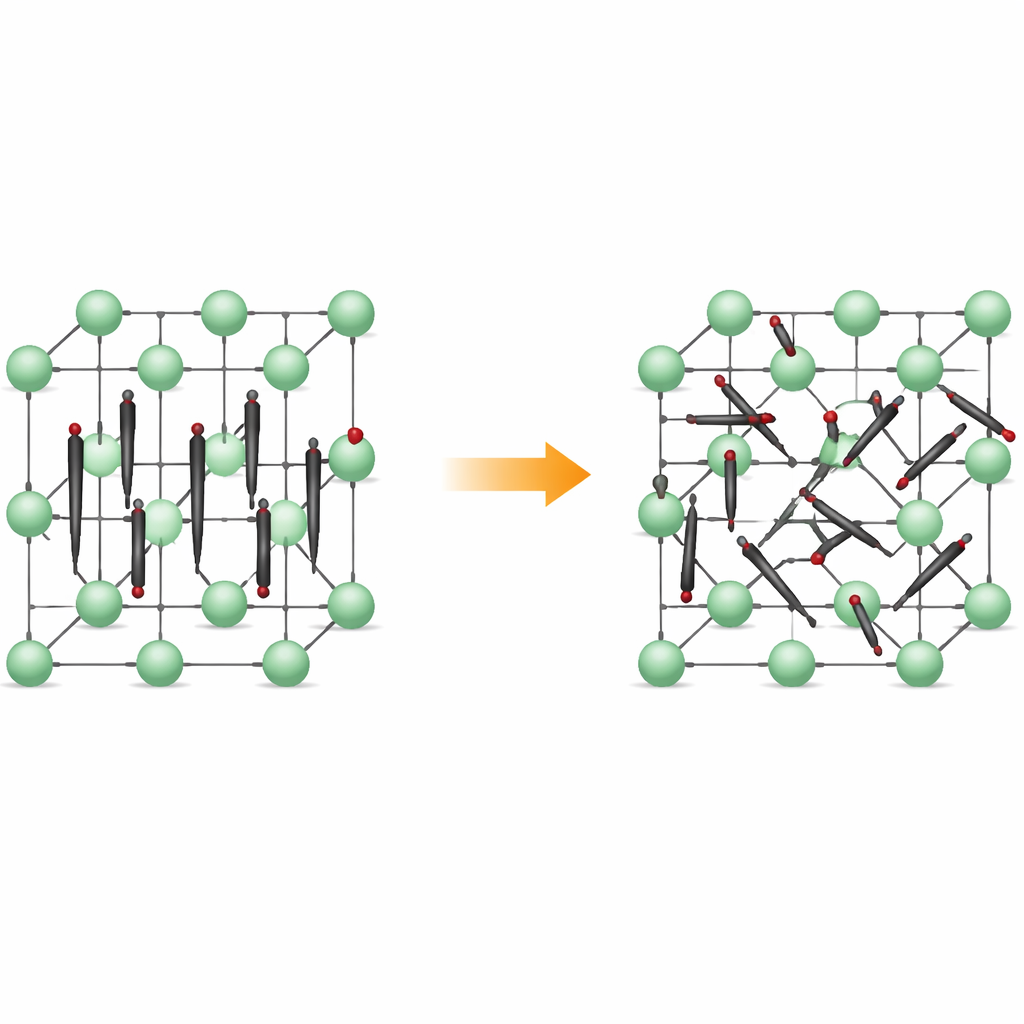
Da file ordinate a un moto irrequieto
A basse temperature, Li2C2 forma un cristallo ortorombico: gli atomi di carbonio si accoppiano in piccoli dimeri C2 che puntano tutti quasi nella stessa direzione, come fiammiferi allineati. Gli ioni litio si trovano negli spazi intermedi, creando un reticolo regolare tridimensionale. Riscaldando il materiale, esso si trasforma in una forma cubica, dove le posizioni dei centri dei dimeri rimangono ordinate su una griglia, ma i dimeri stessi non mantengono una direzione fissa. Invece, ruotano tra diverse orientazioni preferite, trascorrendo tempo in avvallamenti energetici poco profondi corrispondenti a specifici allineamenti. Il materiale resta solido, ma la sua struttura interna diventa dinamicamente disordinata.
Seguire il cambiamento lungo un percorso continuo
Per capire quale fase sia più stabile a una data temperatura, bisogna confrontarne le energie libere, che combinano energia ed entropia (una misura del disordine). I metodi standard basati su piccole vibrazioni attorno a posizioni fisse faticano quando gli atomi si spostano o ruotano in modo significativo. Qui, gli autori usano una tecnica chiamata integrazione termodinamica stress–deformazione, basata su dinamica molecolare ab initio. Costruiscono un percorso di deformazione continuo che rimodella la cella di simulazione dalla struttura ortorombica a bassa temperatura a quella cubica ad alta temperatura. Lungo questo percorso eseguono lunghe simulazioni a temperatura fissata e misurano come lo sforzo interno risponde alla deformazione imposta. Integrando questa risposta di sforzo si ottiene la differenza di energia libera tra le due fasi.
Vedere l’entropia attraverso il moto atomico
I calcoli rivelano che intorno a 600 K la fase ortorombica a bassa temperatura è ancora leggermente favorita, mentre a 650 K la fase cubica prevale con qualche millesimo di elettronvolt per unità di formula. Interpolando questi risultati si ottiene una temperatura di transizione di circa 611 K. Questo valore è inferiore alle stime sperimentali ma rimane in ragionevole accordo, dato l’esiguo scarto di energia libera coinvolto. L’energia interna della fase cubica è in realtà più alta; ciò che la stabilizza è un grande guadagno di entropia, ricondotto direttamente al disordine rotazionale dei dimeri C2. Analizzando come l’orientazione di ciascun dimero perda memoria della direzione iniziale nel tempo, gli autori mostrano che i dimeri si riorientano su scale temporali sub-picosecondo, confondendo il confine tra le categorie usuali di entropia «vibrazionale» e «configurazionale».
Oltre le rappresentazioni semplici del disordine nei solidi
Il lavoro evidenzia anche che scorciatoie comuni — come trattare l’entropia come somma semplice di vibrazioni attorno a configurazioni fisse più un conteggio separato delle orientazioni statiche — falliscono per materiali come Li2C2. Poiché le rotazioni dei dimeri sono rapide e fortemente accoppiate alle vibrazioni ordinarie, il sistema non può essere nettamente separato in parti «vibranti» e «riarrangianti». Il metodo di integrazione stress–deformazione aggira questa difficoltà: estrae l’energia libera completa direttamente dalla dinamica microscopica, senza dover ipotizzare come debba essere ripartita l’entropia.
Cosa ci insegna lo studio
In termini pratici, lo studio mostra come un solido possa rimanere rigido mentre i suoi blocchi costitutivi interni diventano sempre più liberi di torcersi e girare, e come questa libertà interna possa rendere una struttura più disordinata termodinamicamente favorita. Per Li2C2, la fase cubica ad alta temperatura è stabilizzata non perché sia energeticamente più economica, ma perché offre molte più possibilità di orientazione e movimento per i dimeri C2. Dimostrando che l’integrazione termodinamica stress–deformazione può catturare questo sottile equilibrio tra ordine, energia ed entropia, il lavoro apre la strada a prevedere transizioni simili in altri solidi dinamicamente disordinati che potrebbero sostenere futuri dispositivi di raffreddamento, batterie e materiali intelligenti.
Citazione: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
Parole chiave: transizione di fase nello stato solido, disordine dinamico, dinamica molecolare, carburi di litio, integrazione termodinamica