Clear Sky Science · it
Progettazione guidata dai dati di bloccanti LNA per la rimozione efficiente dei contaminanti nelle librerie Ribo-Seq
Perché pulire i dati di sequenziamento è importante
La biologia moderna spesso si basa sulla lettura di milioni di frammenti di RNA per capire come le cellule producono proteine. Ma queste misure potenti, in particolare una tecnica chiamata profilazione dei ribosomi (Ribo‑Seq), possono essere appesantite da frammenti di RNA irrilevanti che sprecano capacità di sequenziamento e denaro. Questo studio descrive un modo semplice, guidato dai dati, per progettare «bloccanti» molecolari specializzati che rimuovono selettivamente quei frammenti indesiderati, quasi raddoppiando l'informazione utile che i ricercatori ottengono dallo stesso esperimento.
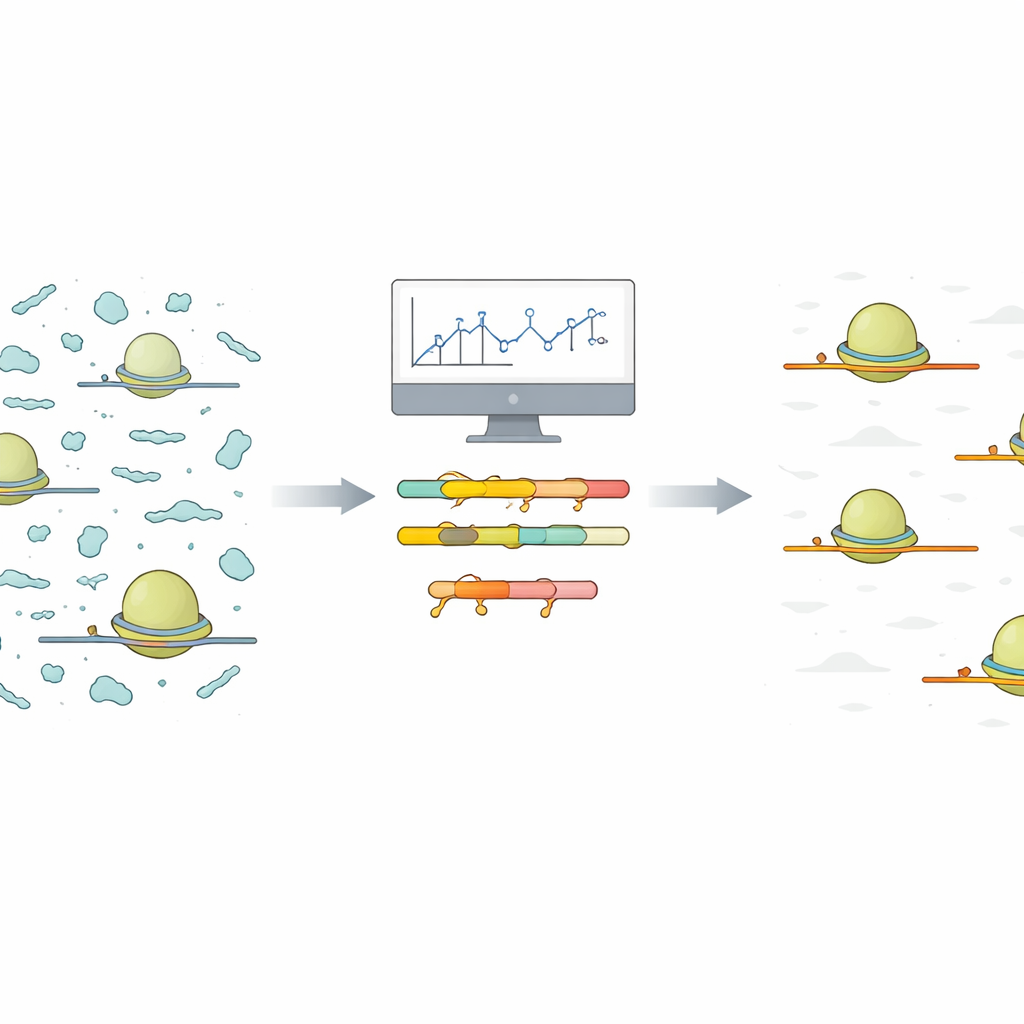
Il problema delle istantanee rumorose dei ribosomi
Ribo‑Seq cattura un'istantanea istante per istante di quali messaggi in una cellula sono attivamente tradotti in proteine. Per farlo, gli scienziati isolano i ribosomi insieme ai brevi tratti di RNA messaggero (mRNA) che essi proteggono. Tutto il resto viene degradato, e i frammenti protetti vengono sequenziati e rimappati sul genoma. In pratica, però, molti altri piccoli pezzi di RNA non codificante passano attraverso questo processo. Poiché questi frammenti contaminanti sono abbondanti e altamente variabili, assorbono una grande frazione delle letture di sequenziamento, lasciando meno letture per i veri segnali codificanti proteine che interessano i ricercatori.
Perché i trucchi di pulizia esistenti non bastano
Le strategie standard cercano di rimuovere RNA ribosomiali abbondanti e altri RNA non codificanti con sonde di cattura preprogettate o enzimi. Questi metodi funzionano bene quando gli RNA target sono intatti e prevedibili, ma Ribo‑Seq taglia intenzionalmente l'RNA in molti frammenti di dimensioni differenti. Questa frammentazione sconvolge i siti target per set di sonde fisse, rendendo la deplezione molto meno efficiente. Inoltre, la miscela esatta di contaminanti dipende dalla specie studiata, dalle condizioni di crescita e persino dall'enzima nuclease utilizzato. I flussi di lavoro di pulizia esistenti tendono anche a coinvolgere più passaggi di incubazione e purificazione, che richiedono tempo e possono causare perdita del campione o bias.
Bloccanti personalizzati progettati a partire da dati reali
Gli autori propongono un approccio snello che inizia con una piccola corsa di sequenziamento pilota a basso costo nelle stesse condizioni previste per l'esperimento completo. Forniscono uno script R che prende le letture allineate di questa corsa pilota e raggruppa automaticamente frammenti contaminanti simili in base alla sequenza. Per ogni gruppo, lo script riporta la sequenza comune più corta presente nei frammenti. Questi brevi tratti condivisi sono siti target ideali per molecole specializzate chiamate oligonucleotidi a acido nucleico bloccato (LNA). Le LNA sono brevi filamenti con una modifica chimica che le fa legare molto strettamente all'RNA complementare. Lo script genera anche mappe di calore intuitive e grafici riepilogativi, aiutando gli utenti a vedere quali contaminanti predominano e quanti target LNA sarebbero necessari per una pulizia sostanziale.
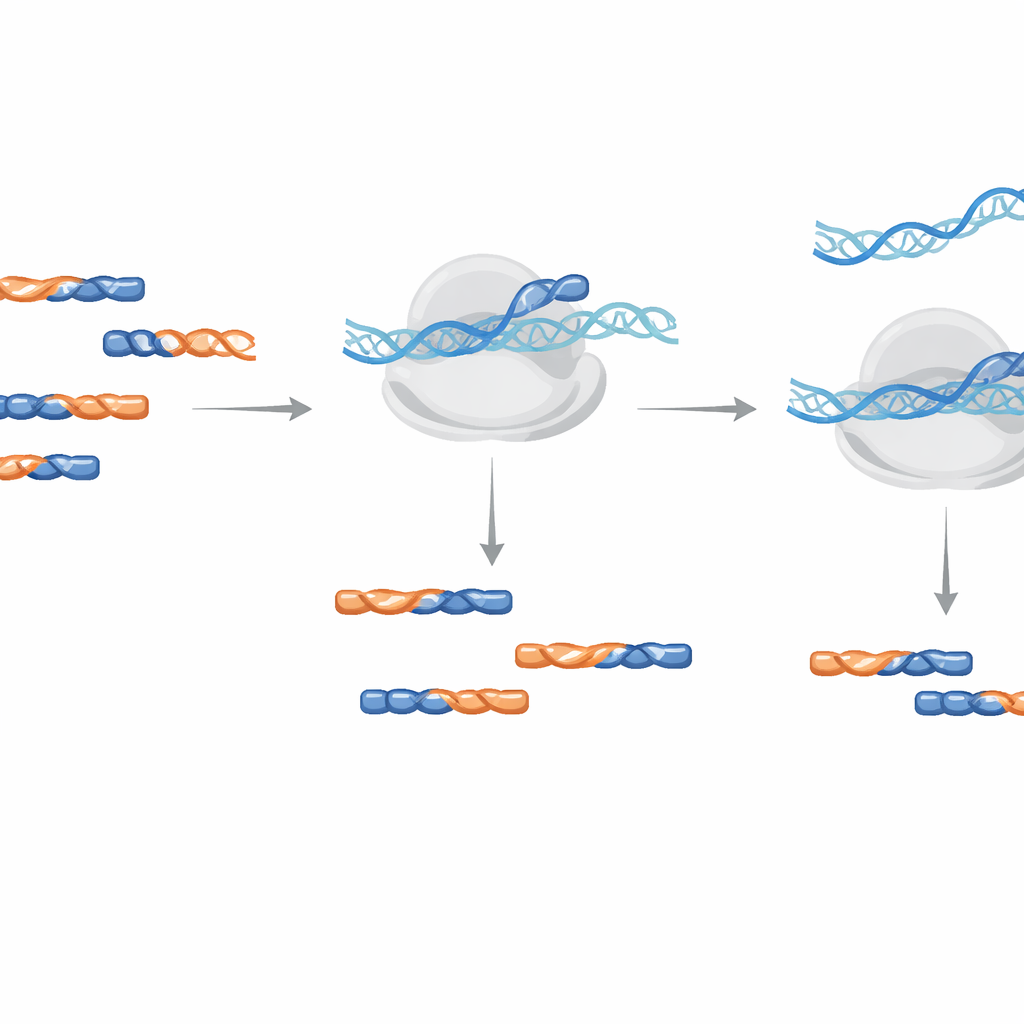
Una pulizia in un unico passaggio durante l'amplificazione
Invece di estrarre fisicamente i contaminanti dal campione, il metodo usa oligonucleotidi LNA come bloccanti durante il passaggio di amplificazione del DNA che costruisce la libreria di sequenziamento. Gli autori hanno testato l'aggiunta di questi bloccanti sia durante la fase iniziale di trascrizione inversa sia durante successiva amplificazione PCR. Hanno scoperto che aggiungere le LNA durante l'amplificazione era più efficiente e richiedeva concentrazioni più basse, riducendo un contaminante di prova di oltre mille volte e funzionando indipendentemente dall'orientamento del filamento. Suggerimenti pratici per la progettazione includono l'alternanza di mattoni standard di DNA e unità LNA, l'uso di una lunghezza minima di 14 unità per la pianta Arabidopsis e la modifica dell'estremità terminale in modo che il bloccante non possa essere esteso accidentalmente.
Piu letture utili senza distorcere il segnale
Per dimostrare le prestazioni nel mondo reale, il team ha progettato cinque bloccanti LNA che prendono di mira i gruppi di contaminanti più comuni osservati nelle condizioni di crescita tipiche di Arabidopsis. Quando hanno aggiunto questa miscela durante l'amplificazione della libreria, la proporzione di contaminanti identificati è diminuita di oltre il 30% e il numero di letture utili codificanti proteine è quasi raddoppiato. Crucialmente, confrontando i conteggi di letture a livello genico tra librerie con e senza trattamento LNA, i valori concordavano quasi perfettamente, indicando che i bloccanti hanno rimosso frammenti indesiderati senza distorcere il segnale biologico proveniente dalle vere impronte di mRNA.
Cosa significa per esperimenti futuri
Questo lavoro mostra che un breve esperimento pilota, unito a uno script di analisi facile da usare e a un piccolo set di bloccanti LNA su misura, può trasformare librerie Ribo‑Seq affollate in dataset molto più puliti e informativi con un unico passaggio di pipettamento. I ricercatori ottengono più letture significative per corsa, risparmiando sui costi e semplificando il disegno sperimentale, pur preservando misure accurate di come i geni vengono tradotti. Gli autori forniscono anche profili di contaminanti e progetti di bloccanti pronti per condizioni vegetali comuni, e suggeriscono che risorse simili potrebbero essere costruite per molti organismi, rendendo la profilazione dei ribosomi di alta qualità più accessibile nella comunità di ricerca.
Citazione: Ricciardi, D.A., Peter, F.E. & Böhmer, M. Data-driven design of LNA-blockers for efficient contaminant removal in Ribo-Seq libraries. Sci Rep 16, 8565 (2026). https://doi.org/10.1038/s41598-026-43117-3
Parole chiave: profilazione dei ribosomi, contaminanti RNA, acidi nucleici bloccati, pulizia delle librerie di sequenziamento, regolazione della traduzione