Clear Sky Science · it
Modello ibrido di deep learning ottimizzato per la diagnosi del cancro al seno mediante dati genetici
Perché è importante per pazienti e famiglie
Il cancro al seno è oggi il tumore più frequentemente diagnosticato nelle donne a livello mondiale, e individuarlo precocemente può fare la differenza tra la vita e la morte. I medici hanno sempre più spesso accesso alle informazioni genetiche di una persona, ma trasformare decine di migliaia di misurazioni geniche in risposte chiare è straordinariamente difficile. Questo articolo descrive un nuovo modello computazionale che interpreta questi complessi schemi genetici per individuare il cancro al seno e prevederne gli esiti con sorprendente accuratezza, offrendo potenzialmente ai clinici un assistente potente per decisioni più precoci e affidabili.
Dai geni ai segnali di allarme
Ogni tumore mammario porta con sé un’impronta molecolare codificata nell’attività di migliaia di geni. Gli autori si sono proposti di costruire un sistema in grado di leggere direttamente questa impronta, invece di basarsi solo su immagini o su una manciata di geni noti come BRCA1 e BRCA2. Hanno lavorato con due delle più grandi risorse pubbliche di genomica dei tumori: la coorte TCGA di cancro al seno, che include l’attività di 17.814 geni in 590 campioni, e lo studio METABRIC, che contiene informazioni genomiche e cliniche per oltre 1.400 pazienti. L’obiettivo era ambizioso: progettare un metodo capace di gestire questo flusso d’informazioni, individuare i segnali più rivelatori e funzionare in modo affidabile anche su gruppi di pazienti completamente separati.
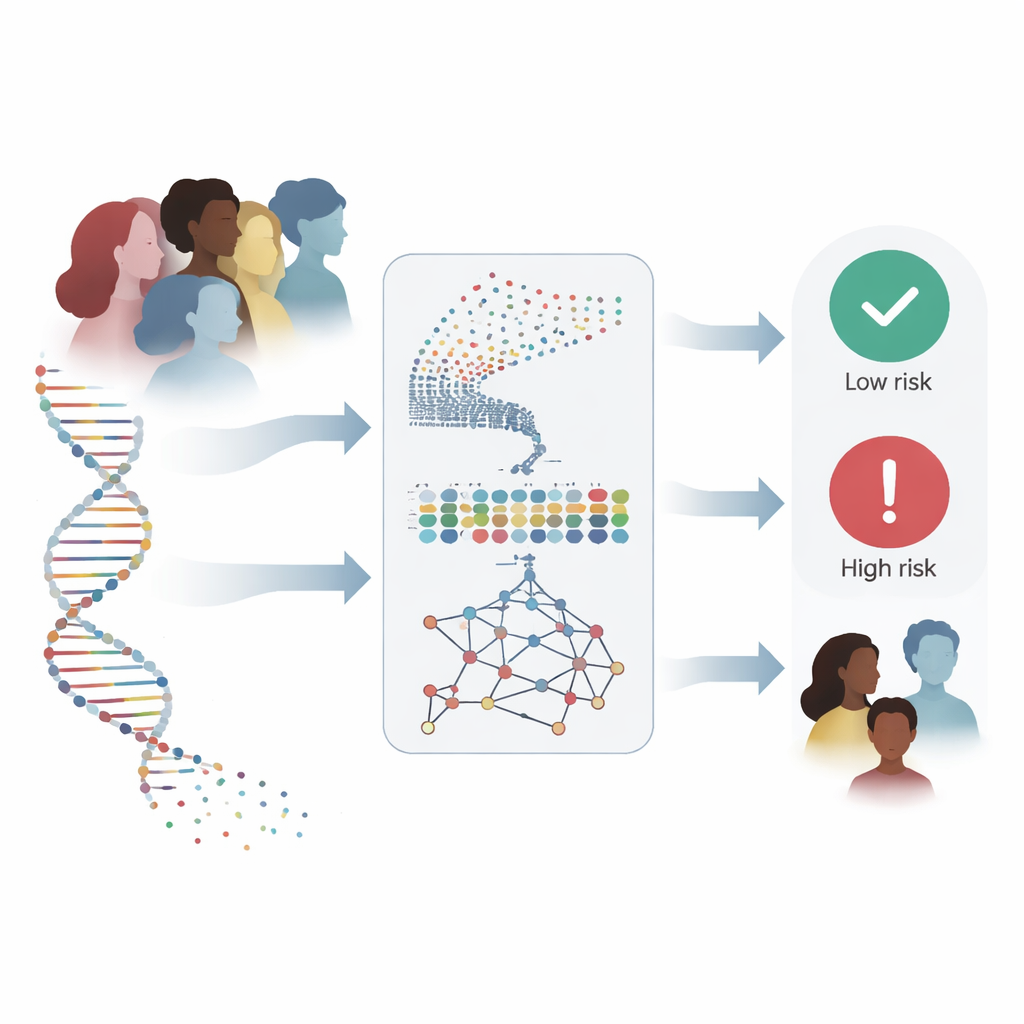
Ridurre migliaia di geni a un insieme utile
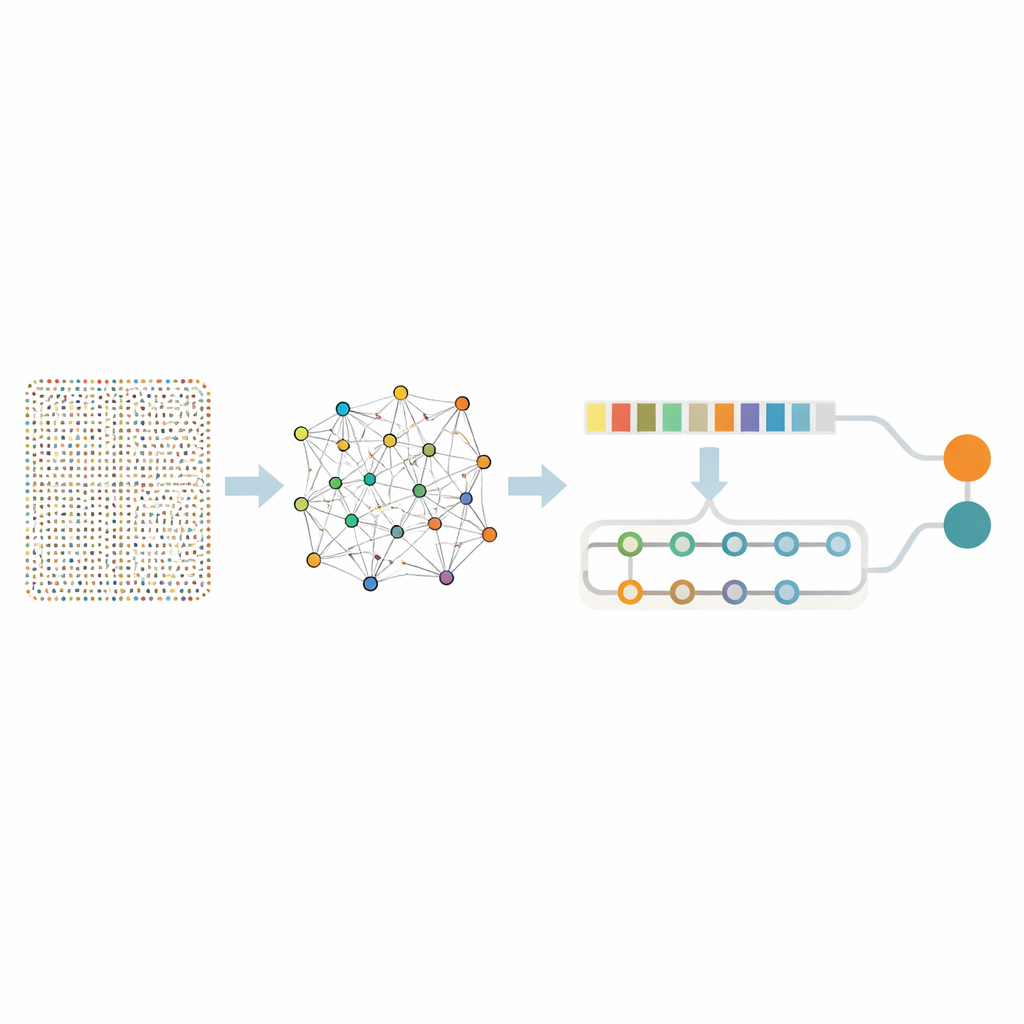
Analizzare quasi diciottomila geni contemporaneamente è opprimente anche per algoritmi avanzati e rischia di catturare solo rumore privo di significato. I ricercatori hanno quindi usato un “setaccio” in due fasi per isolare un sottoinsieme più piccolo di geni realmente informativi. Prima hanno applicato una tecnica chiamata Random Forest, che in pratica interroga molti alberi decisionali su quali geni contino di più per distinguere i tessuti cancerosi da quelli sani. Questo passaggio ha ridotto la lista a 436 geni promettenti. Successivamente hanno esaminato come questi geni si comportano insieme usando l’extraction di regole di associazione, un metodo che individua gruppi di geni che tendono a essere attivi simultaneamente nei tumori. Questo ulteriore livello di analisi ha identificato coppie di geni e reti legate a processi chiave del cancro come la rapida divisione cellulare, la riparazione del danno al DNA e le modificazioni del tessuto circostante il tumore. Dopo questa raffinazione sono rimasti 332 geni: ancora ricchi di significato biologico ma molto più gestibili per analisi approfondite.
Una rete neurale in due parti che apprende pattern e contesto
Con questo set di geni focalizzato, il gruppo ha costruito un modello ibrido di deep learning che combina due tipi di reti neurali. Una parte, nota come rete convoluzionale, scansiona la lista dei geni per cogliere pattern locali—cluster di geni che tendono ad alzarsi o abbassarsi insieme. La seconda parte, una rete a memoria bidirezionale, osserva le stesse informazioni mantenendo traccia delle relazioni a lungo raggio, catturando come geni distanti si influenzino reciprocamente sull’intero profilo. Prima dell’addestramento, gli autori hanno bilanciato i dati in modo che campioni con e senza cancro fossero rappresentati equamente e hanno aggiunto piccole quantità di rumore artificiale, insegnando al modello a non farsi ingannare da fluttuazioni casuali.
Come si comporta il sistema nei test reali
Quando addestrata e testata sui dati TCGA, la rete ibrida ha distinto correttamente i campioni tumorali da quelli normali con circa il 97% di accuratezza e una capacità quasi perfetta di separare i due gruppi. È importante notare che ha superato architetture di deep learning più semplici e strumenti classici di machine learning come la regressione logistica e le macchine a vettori di supporto, anche quando questi metodi concorrenti hanno ricevuto gli stessi geni accuratamente selezionati. La prova più forte è stata però verificarne la robustezza su un dataset completamente diverso. Applicata al METABRIC, raccolto in altri ospedali con metodi di laboratorio differenti, il sistema ha mantenuto alte prestazioni: nella sua migliore esecuzione ha raggiunto il 99,3% di accuratezza e ha identificato correttamente ogni paziente che in seguito è deceduto per cancro al seno, una caratteristica cruciale se lo strumento fosse impiegato per segnalare i casi ad alto rischio.
Cosa potrebbe significare per la cura futura
Per un non specialista, il risultato principale è che questo studio fornisce un filtro e un lettore intelligenti per i dati genetici in grado di individuare il cancro al seno e i relativi rischi con notevole coerenza su ampi gruppi di pazienti. Combinando una strategia accurata di selezione genica con una rete neurale a due rami, gli autori dimostrano che i computer possono estrarre segnali clinicamente significativi da enormi dataset genetici, non solo in uno studio ma attraverso coorti indipendenti. Pur richiedendo ulteriori verifiche su popolazioni diverse e un’analisi più approfondita delle motivazioni delle decisioni, il metodo indica un futuro in cui un semplice campione di sangue o tessuto potrebbe alimentare tali modelli e aiutare i medici a rilevare i tumori prima e a personalizzare i trattamenti con maggiore precisione.
Citazione: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
Parole chiave: genomica del cancro al seno, diagnosi con deep learning, biomarcatori di espressione genica, rilevamento precoce del cancro, supporto decisionale clinico