Clear Sky Science · it
L’apprendimento automatico interpretabile razionalizza l’inibizione della carbonic anidrasi tramite predizione conformale e controfattuale
Perché contano farmaci antitumorali più intelligenti
I farmaci contro il cancro spesso agiscono come strumenti grossolani: pur colpendo le cellule tumorali, possono danneggiare anche i tessuti sani e causare effetti collaterali gravi. Un modo promettente per rendere più mirata questa azione è bloccare versioni specifiche di un enzima chiamato carbonic anidrasi, che aiuta i tumori a sopravvivere in ambienti poveri di ossigeno. Tuttavia, diverse isoforme di questo enzima appaiono quasi identiche, rendendo difficile progettare molecole che colpiscano le isoforme “nocive” nei tumori senza interferire con l’isoforma “buona” presente nell’organismo. Questo studio mostra come l’apprendimento automatico interpretabile possa aiutare i ricercatori a affrontare questa sfida e a progettare candidati farmaceutici più selettivi e sicuri.
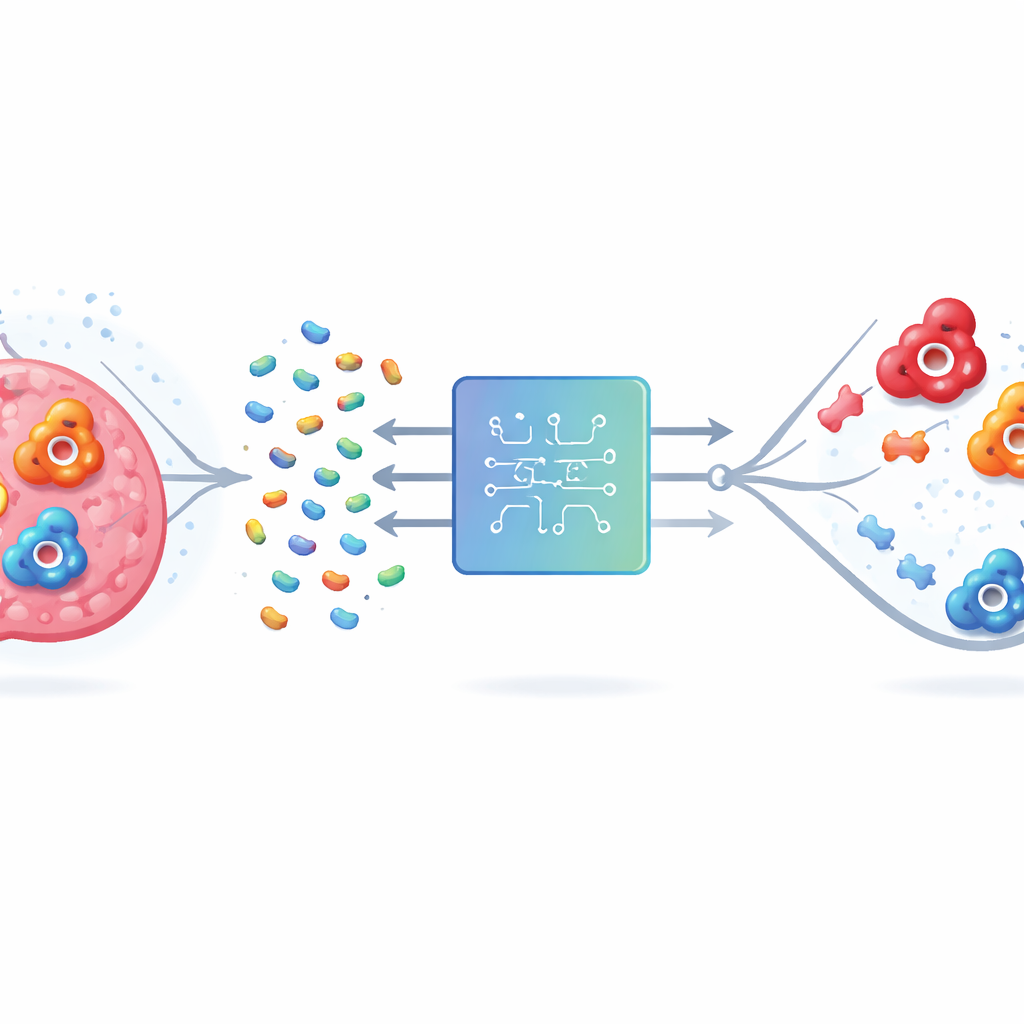
Il problema di colpire il bersaglio sbagliato
La carbonic anidrasi umana (hCA) esiste in molte forme, o isoforme. Due di esse, IX e XII, sono legate alla sopravvivenza delle cellule tumorali in tessuti poveri di ossigeno, quindi il loro blocco potrebbe rallentare la malattia e migliorare il trattamento. Ma l’isoforma II è diffusa nei tessuti sani e presenta un sito attivo molto simile a IX e XII. Farmaci che legano tutte e tre le isoforme possono provocare effetti indesiderati quali acidosi metabolica e disturbi della vista. I metodi tradizionali di laboratorio e computazionali faticano perché gli enzimi sono molecole grandi e complesse e il numero di possibili composti con caratteristiche drug-like è astronomico. Testarli esaustivamente, in laboratorio o in silico, non è semplicemente fattibile.
Costruire una base dati pulita e affidabile
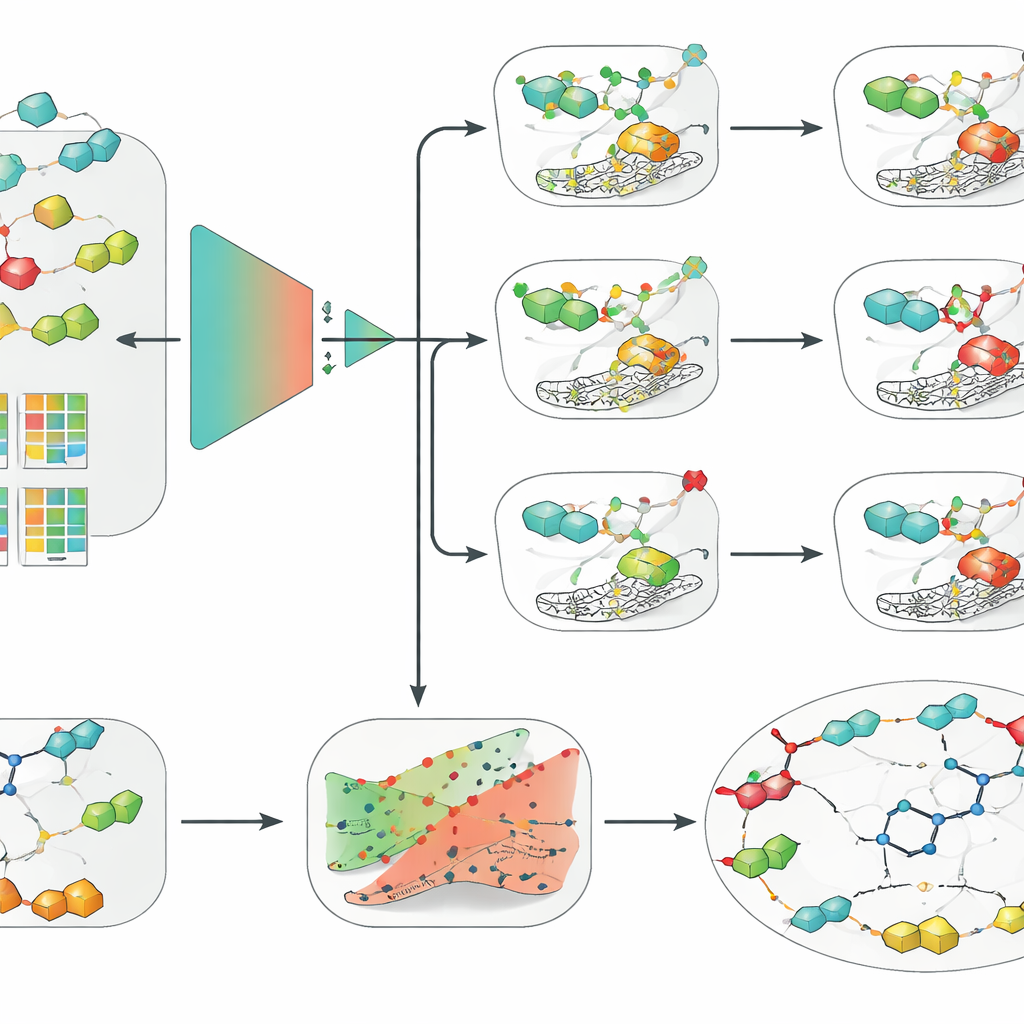
Gli autori hanno affrontato la questione assemblando innanzitutto un database attentamente pulito di migliaia di molecole testate contro hCA II, IX e XII tratte dal repository ChEMBL. Hanno standardizzato le strutture chimiche, rimosso misurazioni dubbie e concentrato l’attenzione su composti che condividono un comune gruppo legante lo zinco tipico di questa classe di inibitori. Usando soglie rigide, hanno etichettato le molecole come chiaramente attive o chiaramente inattive e scartato i casi ambigui che avrebbero potuto confondere i modelli. Poiché le molecole inattive erano molto più numerose di quelle attive, hanno bilanciato i dati in modo che gli algoritmi di apprendimento non favorissero semplicemente la classe maggioritaria. Hanno inoltre adottato una suddivisione basata sullo scaffold in modo che set di addestramento e di test contenessero framework molecolari differenti, offrendo un quadro più realistico di come i modelli si comporterebbero su composti realmente nuovi.
Modelli semplici battono il deep learning quando i dati sono limitati
Con questo dataset curato, il team ha confrontato un’ampia gamma di approcci, da metodi classici di machine learning come regressione logistica, random forest e macchine a vettori di supporto (SVM) a moderne reti neurali profonde, inclusi modelli basati su grafi che operano direttamente sulle strutture molecolari. Li hanno accoppiati con diversi modi di codificare le molecole, come descrittori tradizionali fatti a mano, fingerprint basati su chiavi e embedding appresi da un modello linguistico chimico. Su tutte e tre le isoforme enzimatiche e sotto la valutazione più rigorosa basata sugli scaffold, una combinazione si è distinta costantemente: una SVM alimentata con fingerprint a connettività estesa, un modo strutturato di descrivere gli ambienti chimici locali all’interno di una molecola. Sorprendentemente, questa soluzione relativamente semplice ha superato modelli grafici e di deep learning più alla moda, sottolineando che la qualità dei dati, una validazione accurata e buoni descrittori molecolari possono contare più della complessità algoritmica quando i dataset sono di dimensioni modeste.
Aggiungere fiducia affidabile e spiegazioni comprensibili
I ricercatori hanno quindi avvolto il loro migliore modello SVM in due strati aggiuntivi progettati per rendere le predizioni più utili nella scoperta di farmaci. Innanzitutto, hanno applicato un framework chiamato predizione conformale, che non restituisce solo una risposta sì/no ma fornisce invece un intervallo di esiti probabili insieme a un tasso di errore garantito. Questo permette agli scienziati di regolare quanto cauti vogliono che siano i modelli e di riconoscere i casi in cui il modello è realmente incerto. In secondo luogo, hanno utilizzato spiegazioni controfattuali per rendere il ragionamento del modello più intuitivo. Per una data molecola hanno generato analoghi strettamente correlati che invertissero l’esito predetto da attivo a inattivo, o viceversa. Esaminando queste coppie per il candidato clinico SLC-0111, che blocca selettivamente IX e XII ma non II, il metodo ha riscoperto indipendentemente un’importante intuizione di chimica farmaceutica: piccole modifiche nella “coda” della molecola alterano fortemente quale isoforma preferisce legare.
Dagli algoritmi agli strumenti pratici per il design di farmaci
Per rendere il loro approccio accessibile, gli autori hanno impacchettato i tre modelli SVM, lo strato di incertezza e il motore controfattuale in uno strumento grafico chiamato CAInsight. Un utente può fornire la rappresentazione testuale di una molecola e, con un solo clic, ottenere l’attività predetta contro hCA II, IX e XII, una stima di quanto ogni predizione sia affidabile e suggerimenti di modifiche strutturali che potrebbero aumentare o ridurre l’attività. Pur concentrandosi sulla classificazione delle molecole come attive o inattive piuttosto che sulla previsione di potenza o selettività esatte in un unico passaggio, i modelli riproducono già il comportamento noto per veri candidati farmaceutici e distinguono cambiamenti strutturali sottili. Gli autori osservano che dataset più grandi e più uniformi, oltre a un’analisi più approfondita di come vengono scelti i cutoff di attività, potrebbero perfezionare ulteriormente le prestazioni.
Cosa significa per i futuri farmaci antitumorali
In termini semplici, questo lavoro mostra che modelli di apprendimento automatico costruiti con cura e ben spiegati possono aiutare i chimici a progettare farmaci antitumorali che distinguano meglio tra bersagli enzimatici molto simili. Combinando statistiche robuste, stime di incertezza ed esempi intuitivi di “what-if”, il quadro non solo predice quali molecole hanno più probabilità di funzionare ma suggerisce anche perché. Questo tipo di intelligenza artificiale trasparente potrebbe accelerare lo screening virtuale, supportare la progettazione generativa di nuovi composti e ridurre il carico di tentativi ed errori in laboratorio, contribuendo infine alla scoperta di terapie più selettive e più sicure per i pazienti.
Citazione: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Parole chiave: inibitori della carbonic anidrasi, apprendimento automatico interpretabile, selettività dei farmaci, predizione conformale, spiegazioni controfattuali