Clear Sky Science · it
Mutazioni eterozigoti composte del gene CHAT, una missenso e una variante al sito di splicing, in due fratelli con sindrome miastenica congenita
Quando la respirazione cede senza preavviso
Alcuni bambini sembrano sani alla nascita eppure improvvisamente smettono di respirare durante febbri lievi, richiedendo ventilazione d’emergenza. Per le loro famiglie, gli episodi sono terribili e inspiegabili. Questo studio indaga due fratelli giapponesi di questo tipo e ricostruisce gli attacchi potenzialmente letali di debolezza e apnea (pausa nella respirazione) fino a piccole variazioni in un singolo gene che aiuta i nervi a comunicare con i muscoli. Mettendo insieme indizi clinici, sequenziamento genetico e modellizzazione computazionale delle proteine, i ricercatori mostrano come queste mutazioni probabilmente compromettano un enzima chiave e forniscano ai medici un bersaglio più chiaro per diagnosi e trattamento.
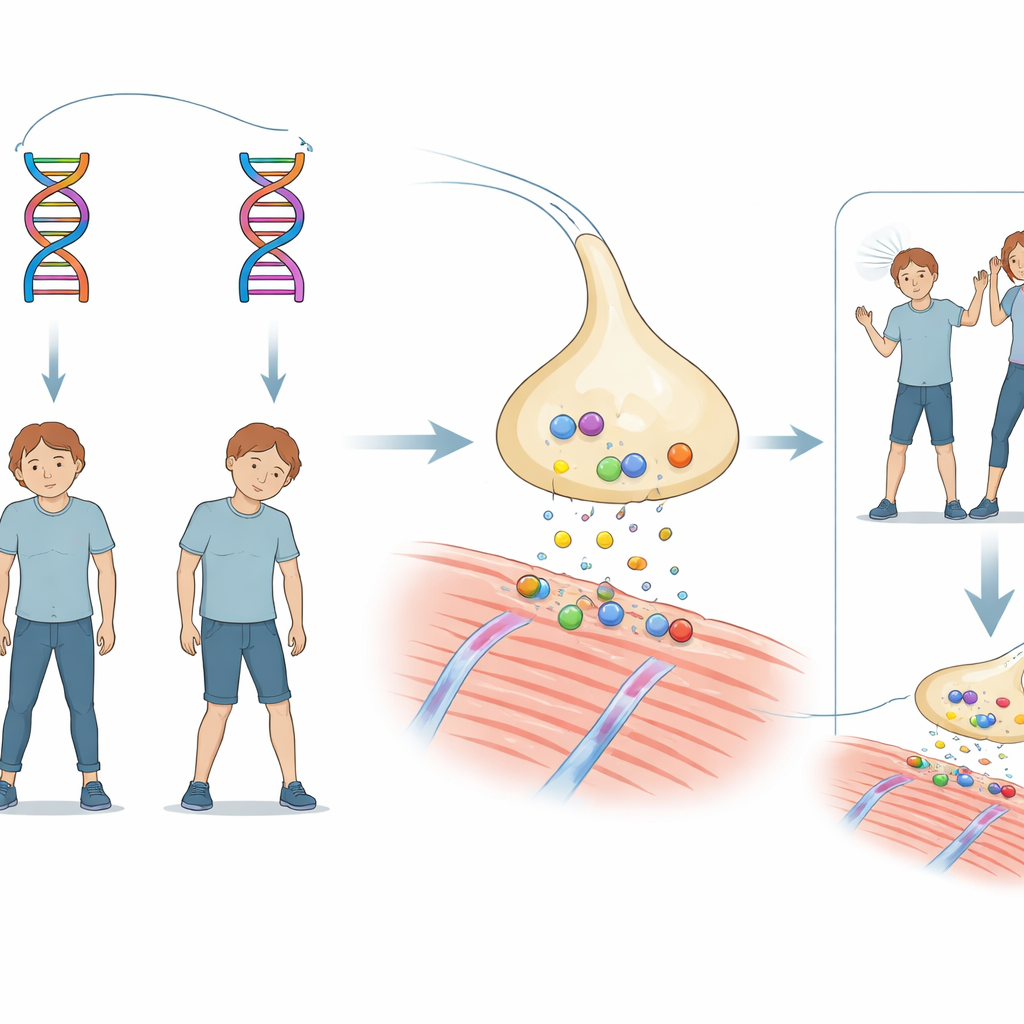
Un mistero familiare di debolezza improvvisa
La storia riguarda un fratello e una sorella che avevano entrambi un leggero ritardo nello sviluppo motorio in età infantile. Intorno ai 18 mesi, ciascuno ha avuto episodi di apnea e perdita di coscienza durante febbri, abbastanza gravi da richiedere un ventilatore. Con la crescita, entrambi hanno continuato ad avere episodi di ptosi palpebrale e debolezza muscolare generalizzata scatenate da infezioni, febbri o sforzo fisico. Le risonanze cerebrali erano normali e sono state escluse le forme più comuni di miastenia mediate da anticorpi (una malattia in cui la comunicazione tra nervo e muscolo è alterata). Tuttavia un farmaco che potenzia il segnale chimico tra nervi e muscoli ha chiaramente migliorato i loro sintomi, indirizzando verso una rara condizione ereditaria chiamata sindrome miastenica congenita.
Trovare le istruzioni difettose
Per cercare una causa ereditaria, il team ha sequenziato tutti i geni codificanti proteine nei fratelli e nei loro genitori. Hanno scoperto che ciascun bambino portava due alterazioni diverse nello stesso gene, CHAT, che codifica per la colina acetiltransferasi—un enzima che produce l’acetilcolina, il principale messaggero chimico usato dai nervi per attivare i muscoli. Un’alterazione ha modificato un singolo mattoncino dell’enzima (una mutazione missenso nota come G411R). L’altra si trovava a un confine critico dove la cellula normalmente taglia e unisce segmenti genici durante la produzione di RNA (una mutazione del sito di splicing etichettata c.752+2T>C). Ciascun genitore portava solo una di queste varianti ed era sano; solo i bambini che avevano ereditato entrambe mostravano la malattia, suggerendo che la coppia di mutazioni insieme indebolisce la funzione enzimatica.
Indagare come un taglio nascosto altera l’enzima
Poiché i ricercatori non sono riusciti a ottenere abbastanza RNA naturale di CHAT dalle cellule del sangue, hanno usato un esperimento con un “minigene”. Hanno clonato il tratto rilevante del gene in un vettore di DNA, introdotto la versione normale o quella mutata in cellule coltivate e poi esaminato come l’RNA veniva processato. Nel costrutto normale, l’RNA conteneva tutti i segmenti previsti. Nella versione mutata, un intero segmento noto come esone 5 veniva saltato, pur mantenendo intatta la cornice di lettura complessiva del gene. Ciò significava che l’enzima sarebbe stato prodotto ma privo di un breve tratto interno di amminoacidi. Confronti evolutivi hanno mostrato che questa regione mancante è altamente conservata tra le specie, suggerendo che svolge un ruolo strutturale importante.
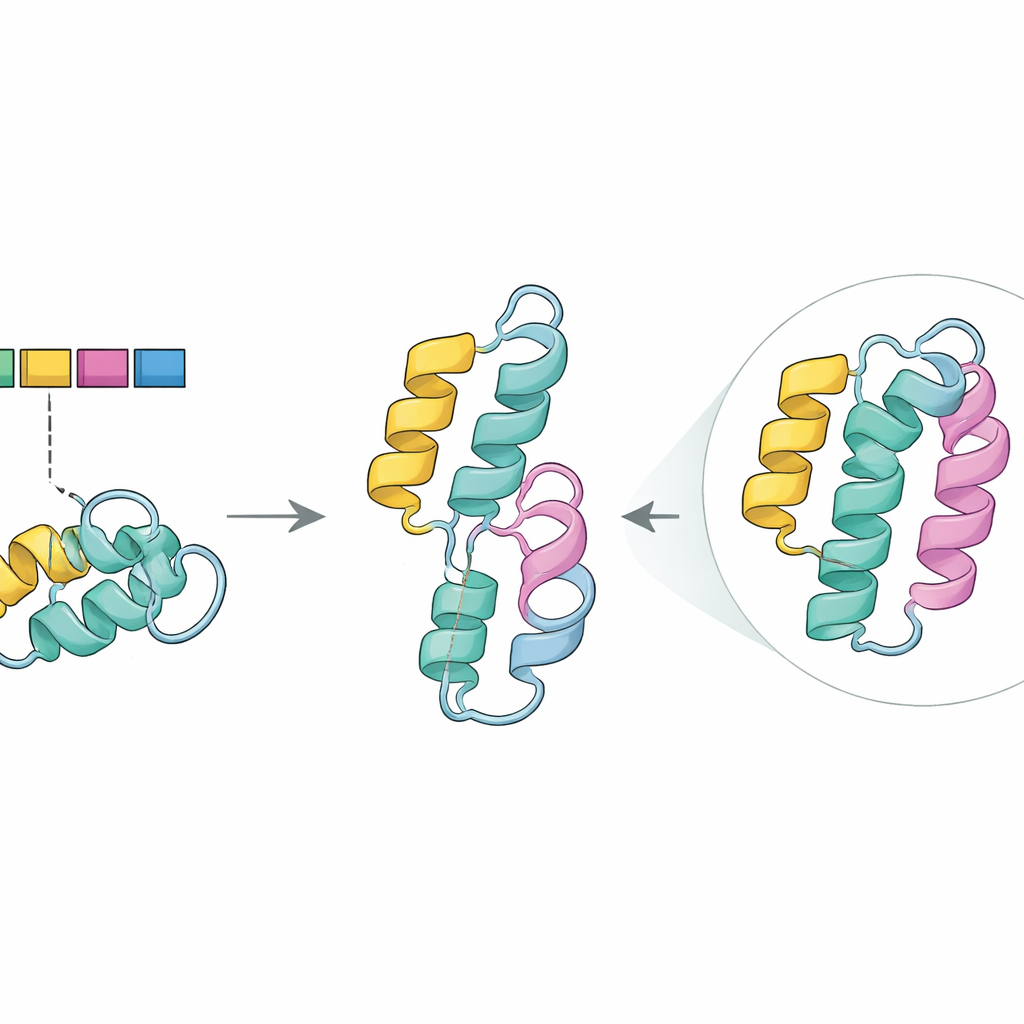
Vedere il danno strutturale in silico
Per esplorare quel ruolo, il team si è rivolto ad AlphaFold2, un programma avanzato che predice le forme tridimensionali delle proteine a partire dalle loro sequenze. Nell’enzima normale, la porzione codificata dall’esone 5 forma uno dei segmenti avvolti strettamente a spirale (un alfa elica) che contribuiscono a stabilizzare il nucleo della proteina. Nella struttura prevista del mutante, questa elica scompariva, lasciando un vuoto in una regione nota da lavori precedenti per essere cruciale nel mantenere la stabilità e nel supportare una chimica efficiente. Insieme a strumenti computazionali che segnalano mutazioni dannose, questi risultati sostengono l’idea che il salto dell’esone 5, specialmente se associato alla variante G411R sull’altra copia del gene, comprometta le prestazioni dell’enzima senza eliminarle completamente—coerente con i sintomi moderati ma gravi dei fratelli.
Cosa significa per pazienti e famiglie
Lo studio conclude che la combinazione della mutazione missenso G411R e della nuova variante del sito di splicing in CHAT è molto probabilmente responsabile della sindrome miastenica congenita nei fratelli. Dimostrando, tramite l’analisi con il minigene e la modellizzazione strutturale, come la mutazione del sito di splicing rimuova un’elica stabilizzante dall’enzima, gli autori forniscono una spiegazione meccanicistica su cui clinici e ricercatori possono costruire. Per le famiglie colpite, questo lavoro offre più di una diagnosi: sostiene trattamenti mirati con farmaci che potenziano la segnalazione neuromuscolare, guida la consulenza genetica per gravidanze future e aggiunge un esempio importante al catalogo di come lievi variazioni nel nostro codice genetico possano influenzare profondamente la forza muscolare e l’atto fondamentale della respirazione.
Citazione: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Parole chiave: sindrome miastenica congenita, gene CHAT, colina acetiltransferasi, mutazione del sito di splicing, giunzione neuromuscolare