Clear Sky Science · it
Deconvoluzione potenziata guidata da attenzione per la stima dei tipi cellulari nello spatial transcriptomics senza riferimento
Vedere le cellule al loro posto
La biologia moderna può leggere l’attività di migliaia di geni contemporaneamente, non solo nelle singole cellule isolate ma direttamente in sottili sezioni di tessuto. Questa visione dello “spatial transcriptomics” rivela dove vivono e interagiscono i diversi tipi cellulari, ma ogni misurazione spesso fonde i segnali di molte cellule vicine. Lo studio presenta un nuovo metodo computazionale, chiamato AGED, che riesce a districare questi mix e stimare quali tipi cellulari sono presenti dove — senza necessitare di un dataset di riferimento single-cell separato e strettamente corrispondente. 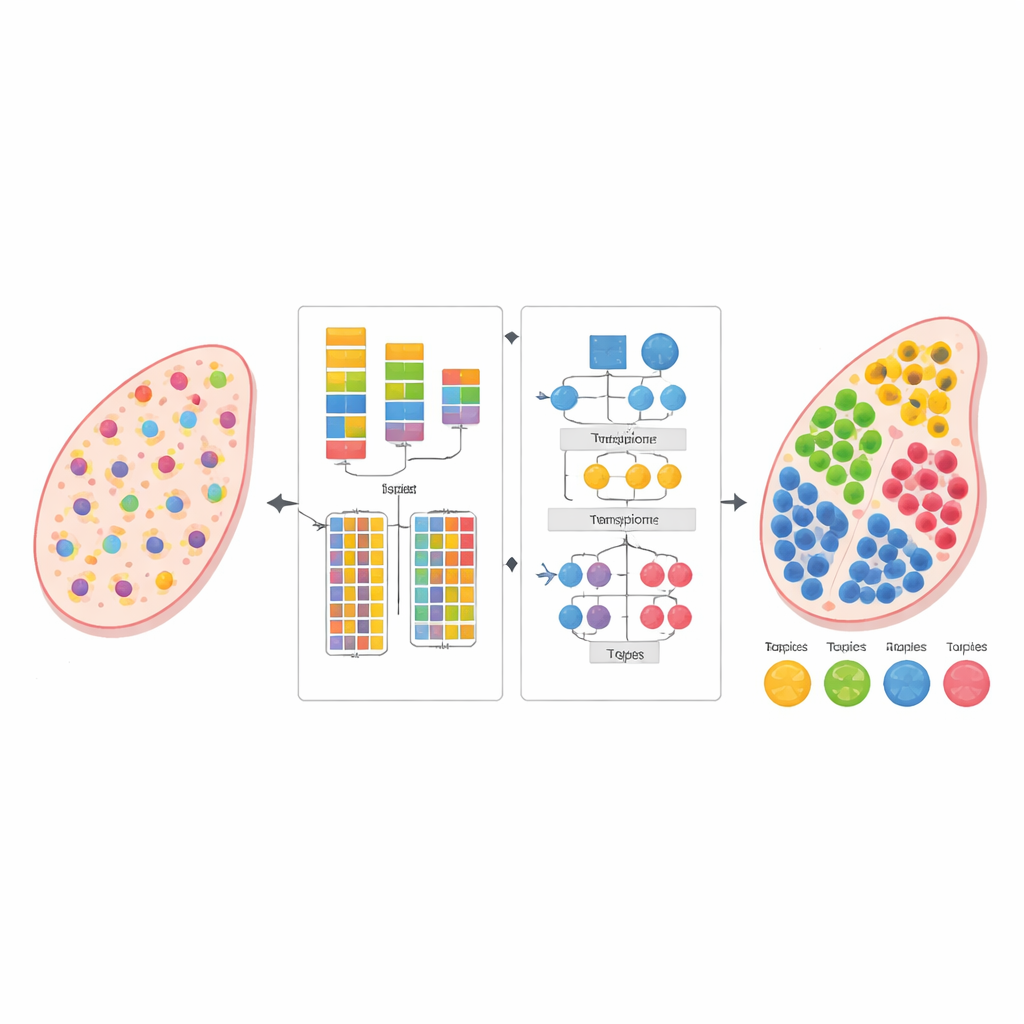
Perché mappare le cellule nei tessuti è difficile
Le piattaforme di spatial transcriptomics misurano l’attività genica su una griglia di spot sovrapposta a una sezione tissutale. Poiché la maggior parte degli spot cattura simultaneamente diverse cellule, i ricercatori devono decomporre matematicamente i segnali misti per recuperare i tipi cellulari sottostanti e le loro proporzioni. Gli strumenti esistenti spesso si basano su atlanti di riferimento single-cell esterni dello stesso tessuto. Quegli atlanti possono mancare per tessuti rari, stati patologici particolari o condizioni sperimentali insolite, e anche quando sono disponibili potrebbero non corrispondere perfettamente, introducendo bias. I metodi senza riferimento evitano questa dipendenza, ma gli approcci attuali faticano con pattern spaziali complessi, relazioni geniche sottili e la sfida di decidere in partenza quanti tipi cellulari distinti cercare.
Una strategia in due fasi per districare i mix
Gli autori hanno progettato AGED come un quadro in due stadi che combina idee dalla statistica e dal deep learning moderno. Nella prima fase, il metodo testa una gamma di possibilità per il numero di tipi cellulari potenzialmente presenti nel tessuto. Utilizza una rete neurale basata sull’attenzione rapida, nota come Performer, per apprendere decomposizioni candidate e poi le valuta con diversi criteri contemporaneamente: quanto bene il modello ricostruisce i conteggi genici osservati, quanto chiaramente i gruppi cellulari inferiti si separano l’uno dall’altro e quanto siano diversi tali gruppi. Una procedura di adattamento di curve individua un “punto gomito” in cui aggiungere altri tipi cellulari porta poco beneficio, permettendo al metodo di selezionare automaticamente un numero adatto invece di affidarsi a una stima dell’utente.
Attenzione guidata per catturare la biologia
Una volta stabilito il numero di tipi cellulari, la seconda fase di AGED affina la soluzione con un’architettura di attenzione più ricca. Parte da un modello statistico di topic che tratta ogni spot tissutale come una miscela di “temi” nascosti — qui a rappresentare i tipi cellulari — e interpreta ogni tipo cellulare come un pattern genico caratteristico. Questi temi iniziali forniscono una struttura globale. Il modello quindi sovrappone diversi meccanismi di attenzione: uno collega i temi statistici alla rete neurale, un altro aggrega informazioni dagli spot vicini nello spazio fisico e un terzo collega direttamente temi e geni. Un sistema di gating permette al modello di decidere, caso per caso, quanto fidarsi dei pattern statistici a priori rispetto ai dati locali. Vincoli aggiuntivi promuovono soluzioni sparse, riflettendo la realtà biologica per cui la maggior parte delle posizioni tissutali è dominata da pochi tipi cellulari principali. 
Mettere il metodo alla prova
I ricercatori hanno valutato AGED su diversi tipi di dati. In tessuto simulato del bulbo olfattivo di topo, il metodo ha recuperato quattro noti strati anatomici e ha ricostruito le composizioni cellulari reali in modo più accurato rispetto a strumenti di riferimento e senza riferimento ampiamente usati, ottenendo sia alta correlazione con il dato di verità sia basso errore di ricostruzione. Nel carcinoma duttale pancreatico umano, AGED ha scelto automaticamente una soluzione a venti tipi cellulari che si è allineata con regioni annotate dal patologo come tumore, dotto e pancreas normale, superando altri metodi su una misura di similarità strutturale che confronta le mappe inferite con la struttura tessutale visibile. Nel tessuto timico umano, AGED ha separato accuratamente popolazioni cellulari chiave e catturato una relazione negativa biologicamente attesa tra due tipi epiteliali specializzati — un pattern che gli approcci concorrenti non sono riusciti a riprodurre. Analisi aggiuntive su altri dataset e a risoluzione simile alla singola cellula hanno ulteriormente supportato la robustezza del metodo.
Cosa significa per il futuro
Per un non specialista, AGED può essere visto come un motore intelligente di demixing per tessuti complessi: impara quanti cluster cellulari distinti sono presenti, dove si trovano e quali geni li definiscono, tutto a partire dai dati spaziali stessi. Intrecciando modelli statistici interpretabili con reti neurali flessibili basate sull’attenzione, il quadro offre sia accuratezza sia intuizione, anche quando non esiste un atlante di riferimento adatto. Questo lo rende uno strumento pratico per esplorare l’organizzazione tissutale in salute e malattia, dai livelli cerebrali ai tumori e agli organi immunitari, e indica una strategia più ampia per usare conoscenze a priori per guidare modelli di machine learning potenti ma opachi in biologia.
Citazione: Yang, X., Wang, Y. & Chen, X. Attention-guided enhanced deconvolution enables reference-free cell type estimation in spatial transcriptomics. Sci Rep 16, 8097 (2026). https://doi.org/10.1038/s41598-026-39703-0
Parole chiave: spatial transcriptomics, deconvoluzione dei tipi cellulari, deep learning, architettura tissutale, analisi senza riferimento