Clear Sky Science · it
Modello di apprendimento analitico guidato dai geni per una diagnosi accurata del tumore al seno
Perché questa ricerca è importante per pazienti e famiglie
Il tumore al seno è oggi il tumore più frequentemente diagnosticato nelle donne a livello mondiale, e pazienti che sulla carta sembrano avere la stessa malattia possono avere esiti molto diversi. Questo studio mostra come i modelli presenti in migliaia di geni, combinati con un sistema di intelligenza artificiale accuratamente progettato, possano aiutare i clinici a distinguere in modo più affidabile chi ha il tumore e quanto possa essere grave—utilizzando solo dati reali dei pazienti e un insieme compatto di geni chiave.
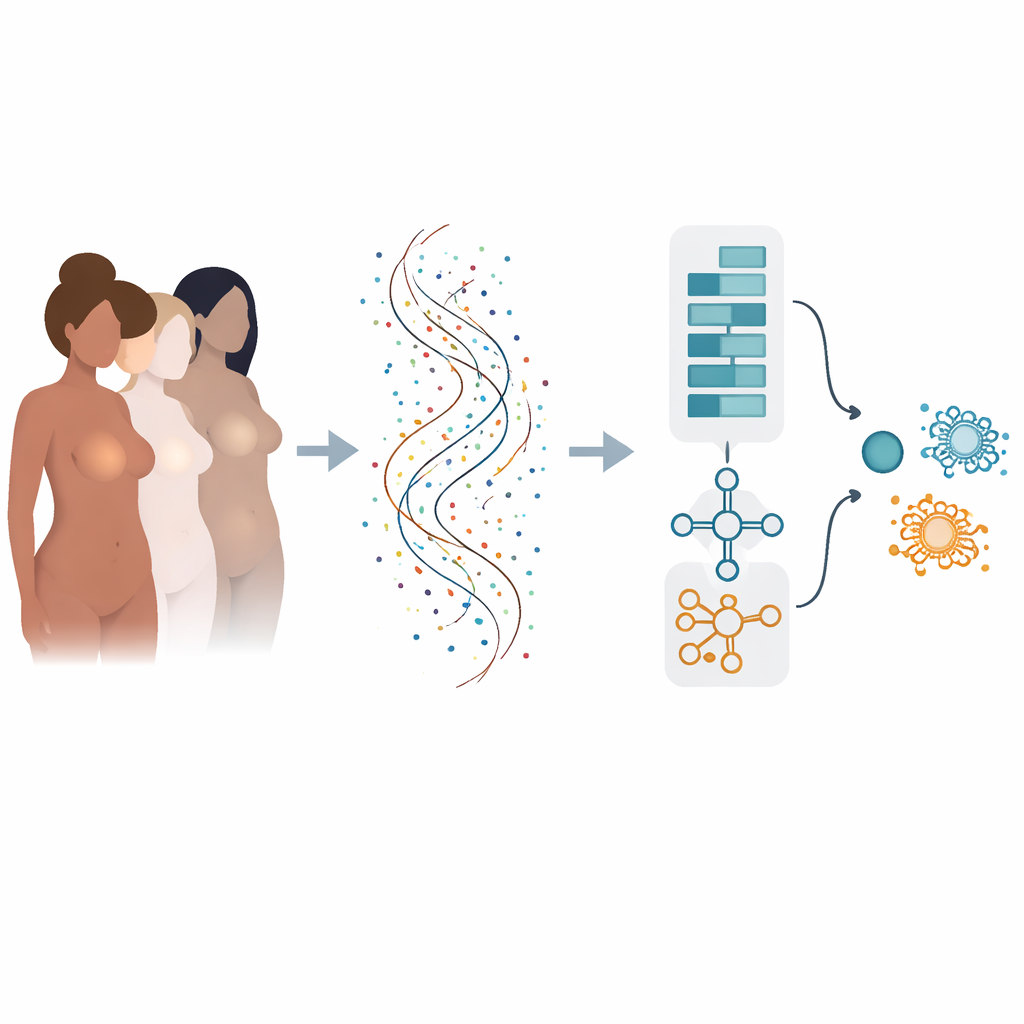
Da molti fattori di rischio al linguaggio dei geni
Il rischio di tumore al seno è modellato da molte influenze: alterazioni genetiche ereditarie, ormoni, peso corporeo, stile di vita e altro. Una volta che il tumore si manifesta, il suo comportamento è determinato da quali geni sono attivi o inattivi all’interno di ciascun tumore. Il sequenziamento moderno può misurare l’attività di decine di migliaia di geni contemporaneamente, ma trasformare questo oceano di numeri in risposte chiare sì/no per diagnosi e prognosi è difficile. I metodi informatici tradizionali spesso esaminano i geni uno per uno e possono perdere il modo in cui gruppi di geni agiscono insieme, oppure possono funzionare bene solo su un dataset e fallire quando testati altrove.
Insegnare a un modello a doppio cervello a leggere i modelli genici
Gli autori hanno costruito un modello di deep learning “ibrido” che agisce un po’ come due cervelli specializzati che lavorano insieme. Una parte, ispirata all’analisi delle immagini, scansiona un elenco ordinato di geni per rilevare pattern locali—cluster di geni la cui attività congiunta segnala il tumore. L’altra parte tratta gli stessi geni come una sequenza, imparando come i geni “driver” iniziali e i geni “a valle” successivi si influenzino reciprocamente lungo l’elenco. Combinando queste due prospettive, il modello può catturare relazioni sia a corto che a lungo raggio all’interno dell’impronta genetica del tumore.
Trovare un nucleo stabile di geni segnali
Invece di inserire tutte le 17.815 varianti geniche misurate nel modello, il team ha progettato una pipeline rigorosa e “senza perdite” per selezionare solo le più informative. Usando una misura standard di correlazione all’interno di cicli ripetuti di controllo incrociato, hanno classificato ripetutamente i geni in base a quanto fortemente la loro attività fosse associata allo stato di tumore. Hanno quindi mantenuto solo i geni che costantemente emergevano nelle posizioni di testa attraverso tutte le suddivisioni di addestramento, ottenendo una firma stabile di 236 geni. I ricercatori hanno anche mappato come questi geni interagiscono tra loro, mostrando che molti formano reti strettamente connesse relative alla crescita tumorale, metabolismo, immunità e ambiente tissutale circostante—evidenza che il gruppo selezionato riflette biologia reale, non rumore casuale.
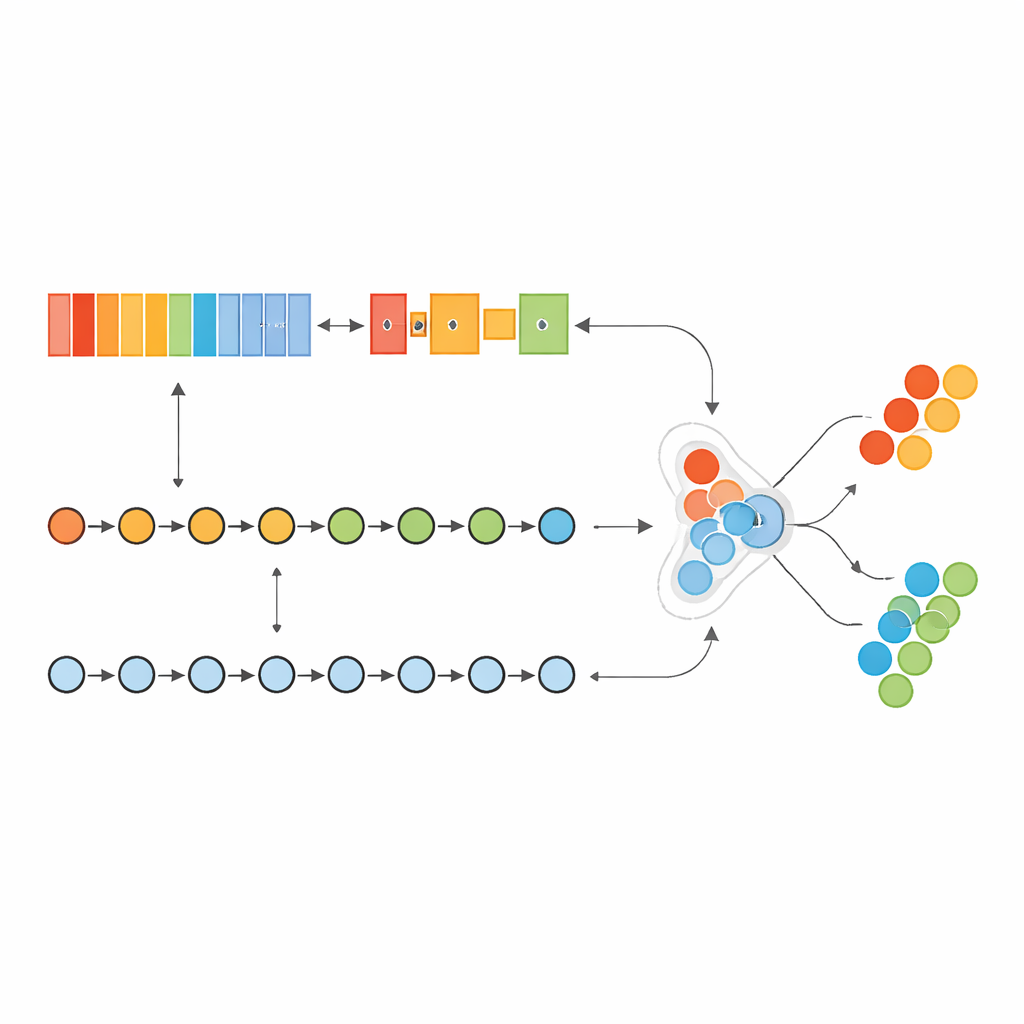
Mettere il modello alla prova
Il sistema ibrido è stato addestrato e ottimizzato su campioni di tumore al seno provenienti da The Cancer Genome Atlas e poi messo alla prova con un dataset completamente separato noto come METABRIC. Per gestire il fatto che i campioni tumorali fossero di gran lunga più numerosi rispetto ai campioni normali, gli autori non hanno creato dati artificiali; invece, hanno regolato quanto il modello "si preoccupasse" degli errori sulla classe più rara. Dopo una ricerca automatizzata dei parametri migliori, il modello ha raggiunto punteggi quasi perfetti sul dataset principale, individuando correttamente quasi tutti i casi di tumore e producendo virtualmente nessun falso allarme. È importante sottolineare che le prestazioni sono rimaste estremamente alte e molto stabili anche quando il modello è stato applicato alla coorte esterna METABRIC, suggerendo che l’approccio può generalizzare oltre uno studio o un ospedale.
Cosa significa per le cure future
In termini semplici, questo lavoro fornisce un’intelligenza artificiale a due componenti finemente sintonizzata che legge un codice compatto di 236 geni per distinguere campioni di seno cancerosi da non cancerosi con sorprendente accuratezza e coerenza, anche in condizioni di rumore. Sebbene lo studio attuale consideri solo l’attività genica e utilizzi dati clinici passati, i suoi metodi pongono le basi per futuri strumenti che potrebbero combinare più tipi di dati—come immagini tissutali e ulteriori livelli molecolari—and fornire spiegazioni chiare su quali geni guidano ciascuna previsione. Con una validazione ulteriore in studi clinici prospettici, un tale sistema potrebbe diventare una spina dorsale universale per la diagnosi di precisione del tumore al seno, aiutando i medici a personalizzare il trattamento usando la “firma” genetica del tumore di ciascun paziente.
Citazione: Hesham, F., Abbassy, M.M. & Abdalla, M. Gene driven analytical learning model for accurate breast cancer diagnosis. Sci Rep 16, 8155 (2026). https://doi.org/10.1038/s41598-026-39430-6
Parole chiave: diagnosi del tumore al seno, espressione genica, deep learning, CNN-BiLSTM, oncologia di precisione