Clear Sky Science · it
CiCLoDS: Clustering cellulare congiunta e selezione genica per trascrittomica spaziale a singola cellula
Trovare i quartieri nella città delle cellule
I microscopi moderni possono ora leggere quali geni sono attivi in centinaia di migliaia di cellule mantenendo però ogni cellula nella sua posizione originale all’interno di un tessuto. Questa rivoluzione della “trascrittomica spaziale” è come trasformare una mappa cittadina sfocata in una vista a livello stradale di ogni casa. C’è però un problema: queste mappe contengono misure per migliaia di geni per cellula, molte più di quante gli scienziati possano facilmente interpretare o permettersi di misurare in esperimenti successivi. Questo studio presenta CiCLoDS, un nuovo metodo che individua quartieri cellulari significativi e, allo stesso tempo, seleziona una lista piccola e interpretabile di geni che definiscono quegli spazi.
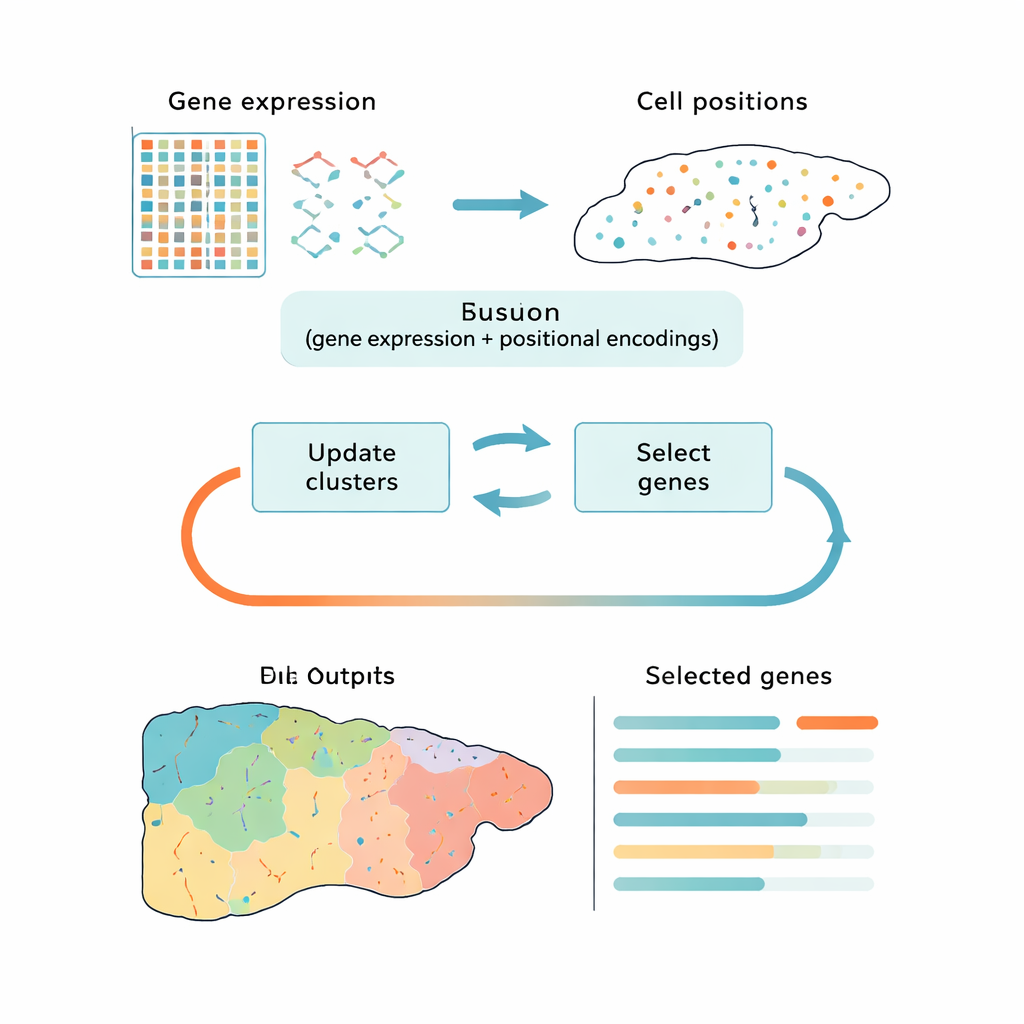
Un modo più intelligente per ridurre i big data
La maggior parte degli strumenti attuali affronta questa sfida in due passi separati: prima riducono i dati a una forma più semplice, poi raggruppano le cellule in cluster. Approcci popolari come l’analisi delle componenti principali (PCA) preservano la variazione complessiva ma possono concentrarsi sul rumore tecnico o su segnali generici del ciclo cellulare piuttosto che sulle differenze biologiche rilevanti. Altri metodi usano l’apprendimento profondo per trovare pattern, ma funzionano spesso come scatole nere e non indicano chiaramente quali geni sono più importanti. CiCLoDS segue una strada diversa. Tratta la selezione genica e il clustering come un unico problema congiunto sotto un “budget” definito dall’utente per il numero di geni da mantenere. In pratica si chiede: quale insieme limitato di geni spiega meglio come le cellule si raggruppano in gruppi distinti, considerando sia la loro attività genica sia, quando disponibile, la loro posizione fisica nel tessuto?
Dalla matematica alle mappe di tessuti reali
Gli autori adattano una famiglia di tecniche matematicamente trasparenti chiamate subspace clustering alle realtà della trascrittomica spaziale, dove i dataset possono contenere oltre un milione di cellule. CiCLoDS lavora su una semplice tabella cellula‑per‑gene, assegnando le cellule ai cluster mentre valuta ciascun gene in base a quanto aiuta a separare quei cluster. Può inoltre integrare informazioni spaziali aggiungendo “codifiche” posizionali che descrivono dove si trova ogni cellula nel tessuto, senza modificare l’ottimizzazione di base. Su grandi dataset di fegato di topo e colon umano generati da piattaforme di imaging ad alta risoluzione, CiCLoDS gira in pochi minuti su computer standard e produce pannelli genici compatti — dell’ordine di alcune decine fino a qualche centinaio di geni — che catturano comunque la ricca struttura dei dati originali.
Rivelare zone nascoste e vasi sanguigni
Applicando CiCLoDS al fegato di topo, il gruppo di ricerca ha verificato se il metodo potesse recuperare i noti schemi di “zonazione” — spostamenti graduali nella funzione degli epatociti da un lato all’altro del lobulo. Rispetto a PCA e a un noto strumento di selezione genica chiamato geneBasis, CiCLoDS ha prodotto zone spaziali più nette con confini più definiti e molto meno regioni assegnate in modo errato, come evidenziato da metriche quantitative che misurano l’accordo con una mappa di riferimento. In modo notevole, quando è stato permesso usare più geni, CiCLoDS ha riscoperto gruppi di epatociti di tipo peri‑portale e peri‑centrale che corrispondevano strettamente a cluster di riferimento definiti da esperti, sebbene non gli fosse stato indicato il biomarcatore chiave AXIN2 né fossero fornite coordinate spaziali esplicite. Quando sono state aggiunte le codifiche spaziali, CiCLoDS ha anche appreso pannelli genici arricchiti per funzioni legate alla superficie cellulare e ai vasi, e ha potuto distinguere con precisione i veri vasi sanguigni dagli artefatti di imaging — qualcosa che metodi più semplici o fallivano nel fare o ottenevano soltanto con aggiustamenti più ad‑hoc.
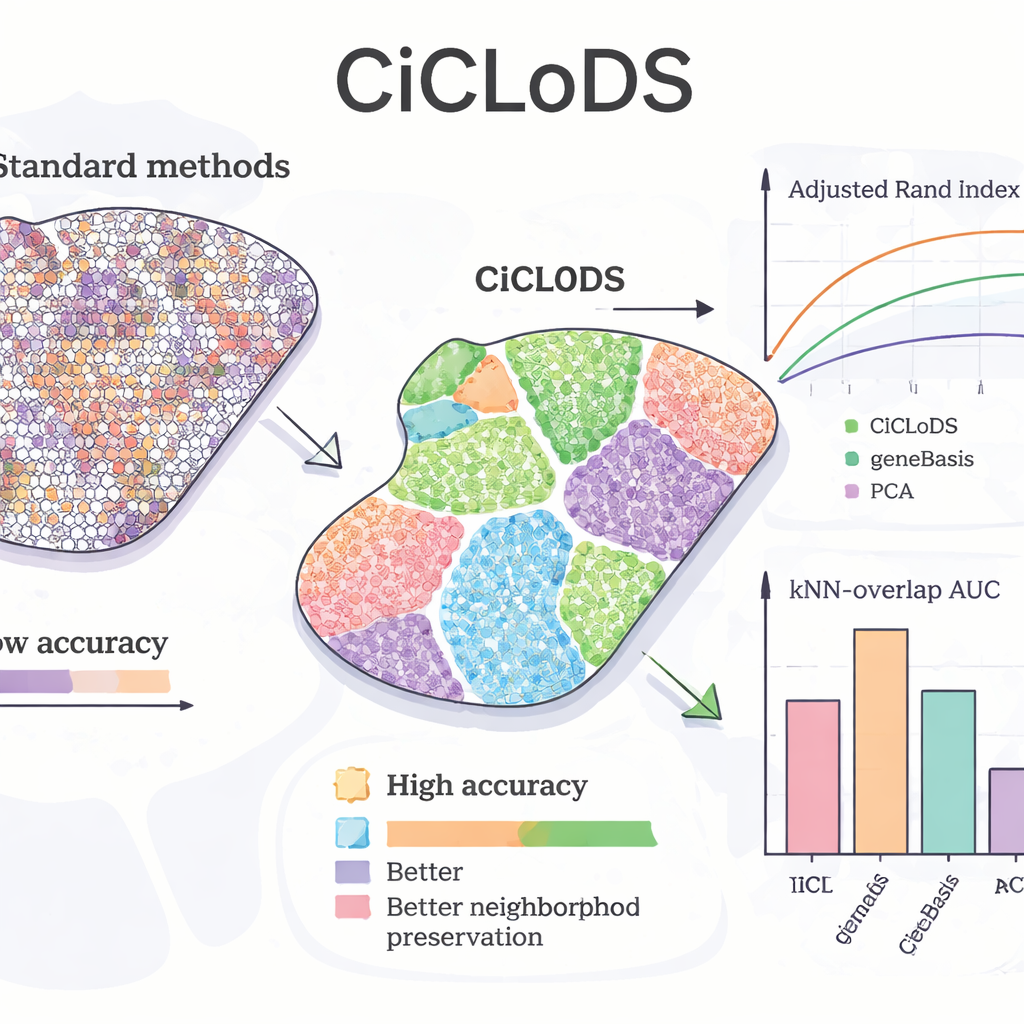
Generalizzare attraverso cervelli diversi e migliorare altri metodi
Per verificare se CiCLoDS regge su tessuti e individui molto diversi, gli autori hanno analizzato campioni della corteccia prefrontale dorsolaterale umana provenienti da tre donatori. Qui, CiCLoDS ha performato alla pari o meglio di metodi spaziali specializzati come BayesCafe e BayesSpace, in particolare su un campione difficile in cui gli altri strumenti hanno faticato. Lo studio mette inoltre in luce un uso “ibrido”: eseguire prima CiCLoDS per ottenere cluster stabili e poi inserirli in BayesSpace. Questa strategia di warm‑start ha aumentato l’accuratezza complessiva e prodotto pattern degli strati cerebrali che corrispondevano meglio alle annotazioni di esperti, dimostrando che CiCLoDS può sia funzionare autonomamente sia rendere i modelli probabilistici successivi più affidabili.
Perché è importante per la biologia e la medicina
Per i non specialisti, la conclusione chiave è che CiCLoDS trasforma mappe cellulari opprimenti in sintesi concise e significative dal punto di vista biologico. Invece di lavorare con migliaia di misure rumorose, i ricercatori ottengono una lista gestibile di geni e cluster spaziali chiari che riflettono l’organizzazione reale del tessuto — zone metaboliche nel fegato, vasi sanguigni e i loro nicchie, e strutture stratificate nel cervello. Poiché il budget di geni è controllato dall’utente e i calcoli sono leggeri, CiCLoDS può aiutare a progettare pannelli genici mirati per esperimenti futuri, guidare l’interpretazione di dataset spaziali complessi e fornire punti di partenza robusti per modelli più elaborati. In un’epoca in cui il collo di bottiglia non è più la raccolta dei dati ma la loro comprensione, strumenti come CiCLoDS promettono di rendere le mappe tissutali ad alta dimensione sia pratiche sia ricche di intuizioni.
Citazione: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
Parole chiave: trascrittomica spaziale, clustering cellulare, selezione del pannello genico, architettura tissutale, analisi a singola cellula