Clear Sky Science · it
Ottimizzazione computazionale della solubilità del dominio calpaia DEK1 tramite modellazione strutturale integrata e mutagenesi mirata guidata dai dati
Perché far comportare le proteine vegetali è importante
Molte delle proteine che regolano la crescita delle piante sono molecole grandi e fragili che rifiutano di dissolversi quando gli scienziati cercano di studiarle in laboratorio. Una di queste proteine, chiamata DEK1, contribuisce a plasmare il corpo della pianta a partire dal livello della singola cellula. Ma poiché una parte cruciale di DEK1 tende a formare aggregati quando viene prodotta nei batteri, la sua struttura tridimensionale è rimasta sconosciuta, rallentando gli sforzi per capirla e sfruttarla. Questo studio mostra come la modellazione al computer e un design intelligente basato sui dati possano riprogettare quella regione problematica per migliorarne la solubilità, senza compromettere la sua architettura—offrendo una ricetta generale per domare proteine ostiche.
Mirare al punto critico in una proteina vegetale chiave
DEK1 è una proteina insolitamente grande inserita nelle membrane cellulari e terminante con una regione enzimatica di taglio nota come dominio calpaia. Lavori genetici hanno dimostrato che questo dominio è essenziale per lo sviluppo normale in piante come i muschi e le colture, eppure la sua struttura non è mai stata risolta sperimentalmente. Quando i ricercatori tentano di produrre questo nucleo calpaico (chiamato CysPc) nel comune ospite batterico Escherichia coli, esso tende a diventare insolubile e a formare corpi di inclusione densi. Ciò rende praticamente impossibile purificarlo nelle quantità e nella qualità necessarie per studi dettagliati strutturali e funzionali. Gli autori hanno quindi deciso di riprogettare il dominio CysPc in modo che si dissolvesse più facilmente pur preservandone la forma complessiva.
Costruire un modello 3D affidabile da zero
Poiché non esiste una struttura sperimentale per questa calpaia vegetale, il gruppo ha prima dovuto prevederne la forma 3D. Hanno combinato diversi strumenti di predizione strutturale all’avanguardia, tra cui AlphaFold2, SWISS-MODEL e I-TASSER, e hanno ancorato queste previsioni a strutture note di calpaine correlate di mammiferi. Usando un approccio di consenso, hanno raffinato e controllato i modelli risultanti con molteplici test di qualità che valutano la geometria della catena principale, il packing e la coerenza con schemi strutturali noti. Questi controlli indipendenti hanno mostrato che il modello integrato del dominio CysPc era più affidabile di qualsiasi singola previsione, fornendo un punto di partenza solido per esplorare come piccole modifiche alla sequenza aminoacidica potessero migliorare la solubilità.
Testare mutazioni virtuali in un solvente simulato
Con il modello 3D a disposizione, gli autori hanno eseguito estese simulazioni di dinamica molecolare, in cui la proteina e le molecole d’acqua circostanti vengono seguite nel tempo al computer. Si sono concentrati su residui sulla superficie della proteina che erano flessibili, idrofobici o predetti promotori di aggregazione. Posizioni candidate sono state mutate singolarmente in aminoacidi più amici dell’acqua e poi simulate per 200 nanosecondi ciascuna. Per ogni variante hanno misurato caratteristiche correlate alla solubilità, come quanta superficie è a contatto con l’acqua, quanto compatta rimane la proteina e quanto oscillano gli atomi. Molte singole mutazioni hanno aumentato modestamente l’esposizione al solvente o il legame idrogeno interno pur lasciando intatta la piega complessiva, suggerendo che l’impalcatura di base del CysPc poteva tollerare sostituzioni scelte con attenzione.
Lasciare che gli algoritmi esplorino lo spazio delle mutazioni
Cambiare un solo residuo raramente produce guadagni drastici in solubilità, quindi i ricercatori hanno esplorato combinazioni di due e tre mutazioni. Hanno generato una libreria di varianti doppie e triple costruite a partire dalle migliori singole mutazioni e hanno simulato ciascuna di esse. Per valutare e classificare equamente questi design, hanno definito un indice pesato che combina più caratteristiche di simulazione note per correlare con la solubilità, premiando un’idratazione aumentata e maggior legame interno e penalizzando un’eccessiva flessibilità. Hanno poi usato un algoritmo di apprendimento per rinforzo (Proximal Policy Optimization) per navigare l’enorme spazio dei possibili tripli mutanti e proporre le combinazioni più promettenti. Questa ricerca guidata dai dati ha convergerto su un particolare triplo mutante, denominato MUT347, come candidato di punta.
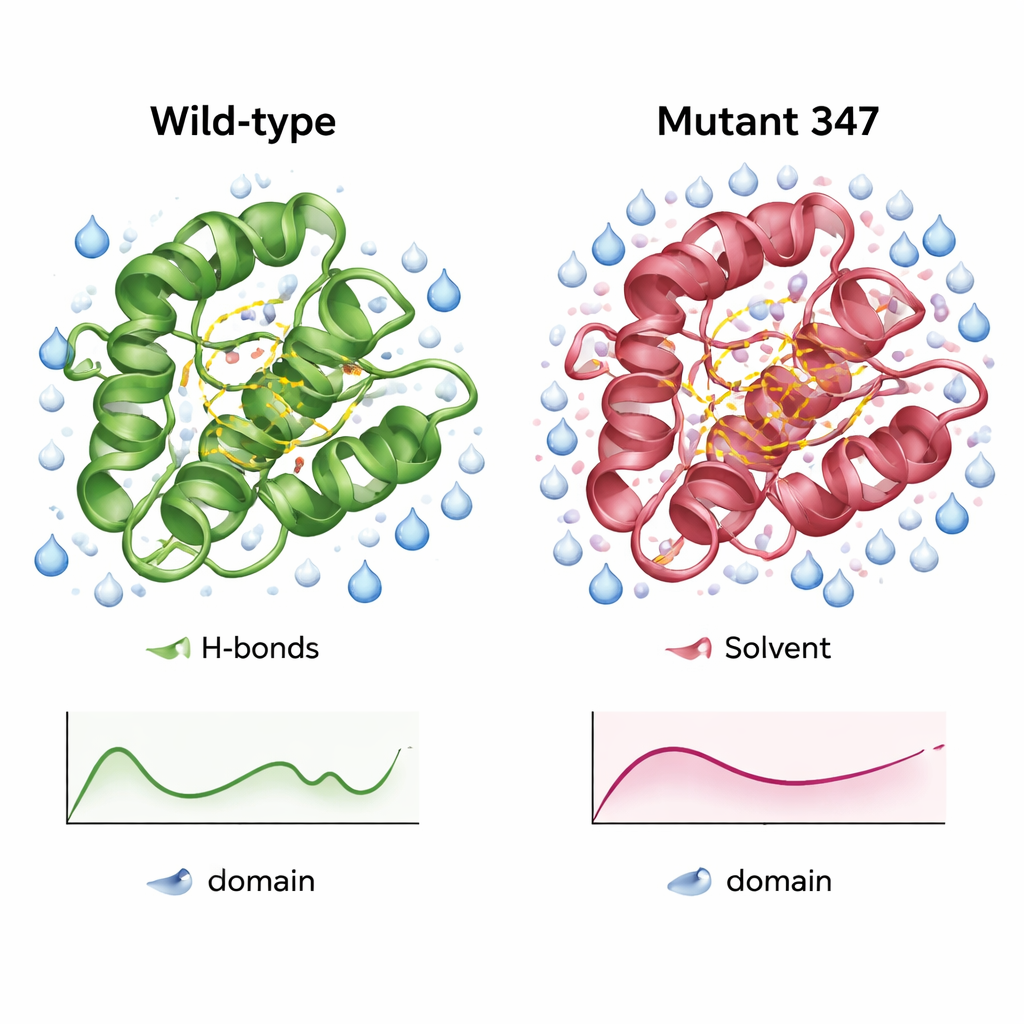
Una versione dell’enzima più compatta e meglio idratata
Simulazioni dettagliate del dominio CysPc selvatico e di MUT347 hanno rivelato come la variante ingegnerizzata differisse. MUT347 si è equilibrata più rapidamente e ha mostrato deviazioni complessive minori rispetto alla forma iniziale, indicando una maggiore stabilità strutturale in soluzione. I suoi loop e le estremità della catena erano leggermente meno mobili, mentre la regione catalitica centrale manteneva la flessibilità originale, suggerendo che i movimenti funzionalmente importanti erano preservati. Il triplo mutante presentava più legami idrogeno interni e una superficie accessibile all’acqua maggiore in regioni chiave, segni di una superficie meglio organizzata e più idratata. A diverse concentrazioni saline e livelli di pH, MUT347 ha mantenuto costantemente fluttuazioni inferiori rispetto alla proteina originale, un comportamento associato a una minore tendenza a formare aggregati.
Cosa significa per lo studio e il riutilizzo delle proteine
Per i non specialisti, il risultato è che gli autori hanno messo a punto una ricetta in gran parte basata su computer per trasformare un frammento difficoltoso e aggregante di una proteina vegetale vitale in una versione più solubile e ben comportata, senza dover conoscere in anticipo la sua struttura sperimentalmente. Combinando predizione strutturale moderna, simulazioni a lungo termine e algoritmi di apprendimento capaci di gestire molte scelte progettuali contemporaneamente, hanno identificato una tripla mutazione che si prevede stabilizzi la piega ed esponga la superficie in modo più favorevole all’acqua. Pur richiedendo ancora conferme sperimentali in provetta, questo quadro potrebbe essere ampiamente utile per recuperare altre proteine eucariotiche difficili da produrre, aiutando in ultima analisi gli scienziati a sbloccare strutture e funzioni attualmente fuori portata.
Citazione: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Parole chiave: solubilità delle proteine, mutagenesi computazionale, dynamica molecolare, calpaia vegetale DEK1, ingegneria delle proteine