Clear Sky Science · it
Il lncRNA FTX promuove la fibrosi miocardica spugnando miR-335-3p per regolare la segnalazione TFEC/ILK
Perché la cicatrizzazione del cuore conta
L’insufficienza cardiaca colpisce decine di milioni di persone nel mondo e spesso si sviluppa silenziosamente nell’arco di anni. Un importante motore di questo declino è la fibrosi miocardica—la lenta e progressiva formazione di tessuto cicatriziale nel muscolo cardiaco che lo rende più rigido e meno efficiente nel pompare il sangue. Questo studio indaga il “cablaggio” molecolare che istruisce le cellule cardiache a depositare troppo tessuto cicatriziale e identifica una nuova catena di molecole che potrebbe essere bersaglio per rallentare o persino invertire questo processo dannoso.
Uno sguardo più ravvicinato alla cicatrizzazione cardiaca
Quando il cuore è lesionato o stressato, cellule di supporto chiamate fibroblasti cardiaci entrano in azione. Nella riparazione sana aiutano a rattoppare i danni. Ma nelle malattie croniche possono trasformarsi in uno stato iperattivo, producendo collagene in eccesso e altri componenti della matrice extracellulare, irrigidendo infine la parete cardiaca. I ricercatori hanno usato due modelli per studiare questo processo: topi trattati con isoproterenolo, che induce in modo affidabile la fibrosi cardiaca, e fibroblasti cardiaci umani esposti alla molecola di segnalazione TGF-β1, un noto attivatore della cicatrizzazione. In entrambi i sistemi hanno misurato come geni e proteine specifici cambiassero durante lo sviluppo della fibrosi.
Una reazione a catena dannosa all’interno delle cellule
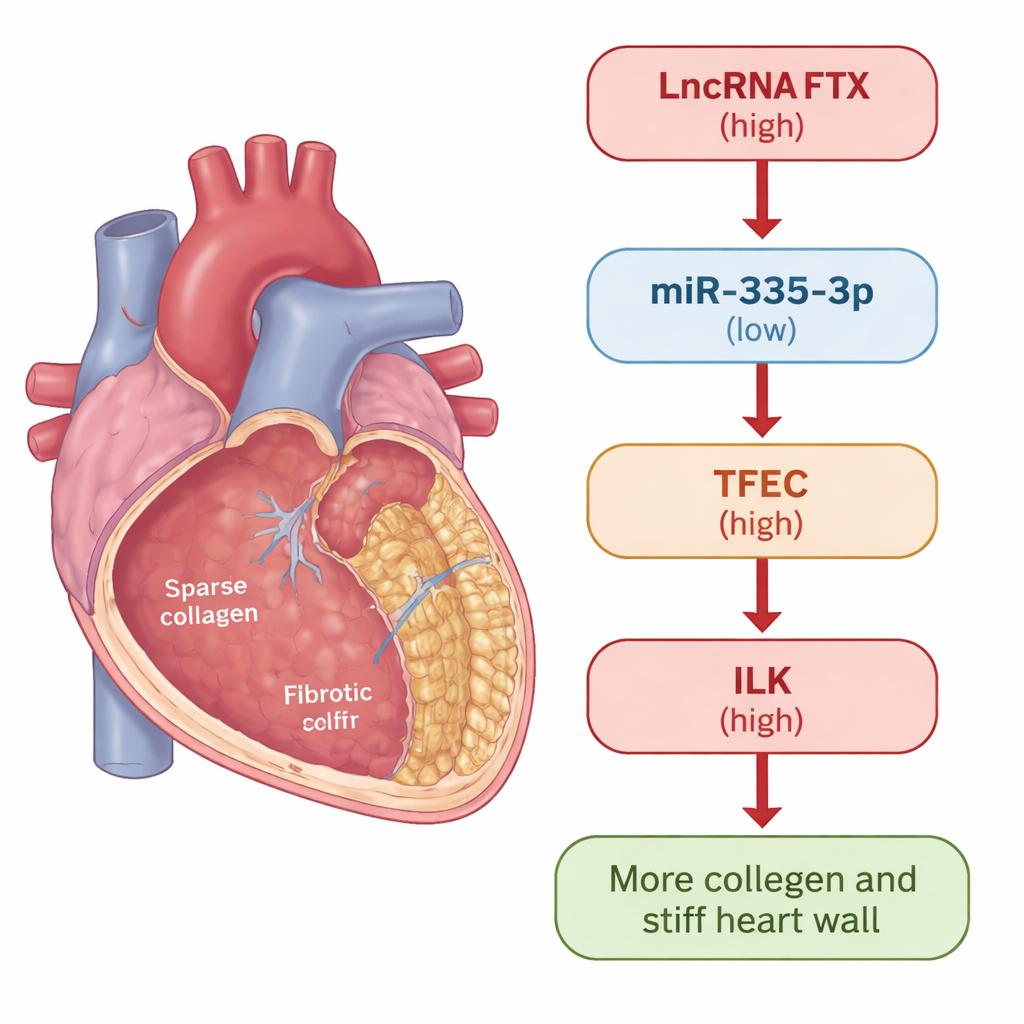
Il gruppo si è concentrato su un fattore di trascrizione chiamato TFEC, una proteina che risiede nel nucleo e attiva altri geni. Hanno osservato che TFEC, insieme a un’altra proteina denominata integrin-linked kinase (ILK), aumentava costantemente quando i fibroblasti venivano indotti verso uno stato fibrotico e produttore di cicatrice. Silenziare TFEC o ILK riduceva nettamente i marcatori classici della fibrosi come l’actina α-smooth muscle e i collageni I e III, e attenuava anche una via di controllo della crescita (Akt/GSK3β e la segnalazione Hippo) nota per favorire la cicatrizzazione tissutale. Esperimenti di mappatura del legame al DNA hanno mostrato che TFEC si attacca direttamente al promotore del gene ILK e ne aumenta l’attività, collocando TFEC chiaramente a monte di ILK in una cascata di segnalazione pro-fibrotica.
Interruttori di RNA che controllano il regolatore principale
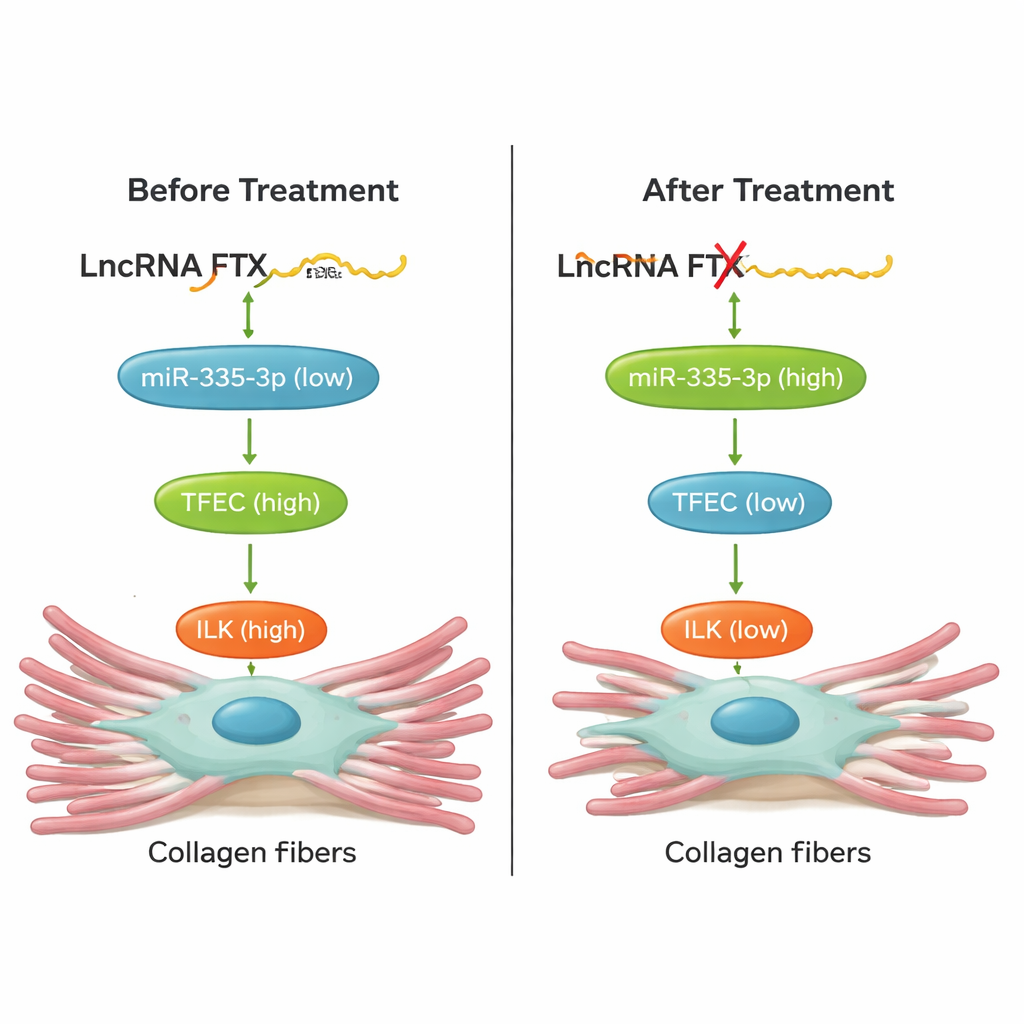
Per capire cosa controlla TFEC gli autori si sono rivolti agli RNA non codificanti—molecole di RNA che non producono proteine ma agiscono come regolatori fini dell’attività genica. Hanno identificato un piccolo RNA, miR‑335‑3p, che risultava ridotto nei cuori e nelle cellule fibrotiche. Aumentare i livelli di miR‑335‑3p abbassava TFEC, mentre bloccarlo ne aumentava l’espressione; test reporter hanno confermato che miR‑335‑3p si lega direttamente ai messaggi di TFEC per mantenerli sotto controllo. Hanno poi individuato un lungo RNA non codificante, chiamato FTX, che era elevato nella fibrosi e interagiva fisicamente con miR‑335‑3p. FTX fungeva da spugna molecolare: assorbiva miR‑335‑3p, impedendo a questo piccolo RNA di limitare TFEC. Di conseguenza TFEC e ILK aumentavano e i fibroblasti producevano più collagene pro-cicatrice.
Dalla coltura cellulare ai cuori vivi
Elemento cruciale, il team ha testato se interrompere questa catena potesse effettivamente proteggere i cuori negli animali. Nei topi esposti all’isoproterenolo, ridurre TFEC, abbattere FTX nel cuore con un vettore di terapia genica AAV9, o aumentare miR‑335‑3p con un “agomir” chimicamente stabilizzato hanno tutti portato a minore accumulo di collagene e a livelli inferiori di marcatori di fibrosi nel tessuto cardiaco. Questi interventi hanno anche migliorato la funzione cardiaca: il volume sistolico e la frazione di eiezione si sono spostati verso valori normali e gli aumenti dannosi della frequenza cardiaca sono stati attenuati. Esperimenti di recupero nelle cellule hanno mostrato che modificare un componente dell’asse FTX/miR‑335‑3p/TFEC/ILK spostava prevedibilmente gli altri, confermando che si tratta di una via strettamente collegata e non di una semplice correlazione.
Cosa significa per i trattamenti futuri
Per chi non è specialista, il messaggio principale è che gli autori hanno identificato una nuova “leva di controllo” per la cicatrizzazione cardiaca. Un lungo RNA chiamato FTX toglie i freni (miR‑335‑3p) da un interruttore maestro (TFEC), che a sua volta attiva ILK e segnali pro-cicatrizzazione a valle, guidando il deposito eccessivo di collagene e l’irrigidimento del cuore. Riducendo FTX, ripristinando miR‑335‑3p o bloccando direttamente TFEC è stato possibile nei topi ridurre la fibrosi e migliorare la funzione di pompaggio. Pur servendo ulteriore lavoro per confermare questa via nei pazienti umani e sviluppare terapie sicure, questa catena regolatoria basata su RNA offre diversi punti promettenti per intervenire nell’insufficienza cardiaca guidata dalla fibrosi.
Citazione: Yao, F., He, Z., Zheng, C. et al. LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling. Sci Rep 16, 7340 (2026). https://doi.org/10.1038/s41598-026-38615-3
Parole chiave: fibrosi miocardica, insufficienza cardiaca, RNA non codificante, fibroblasti cardiaci, segnalazione della fibrosi