Clear Sky Science · it
Approcci integrati per esplorare i cambiamenti temporo-spaziali nel riassortimento genico del virus dell’influenza aviaria altamente patogena A(H5) in Eurasia, 2000–2023
Perché le «zone di remix» genetico dell’influenza aviaria ci riguardano
L’influenza aviaria non è più solo un problema per galline e anatre nelle fattorie lontane. Una forma altamente pericolosa di influenza aviaria, nota come H5, si sta diffondendo in Europa e Asia da più di due decenni, uccidendo uccelli selvatici, sterminando allevamenti di pollame e occasionalmente infettando mammiferi, tra cui bovini e esseri umani. Questo studio pone una domanda semplice ma urgente: dove e in quali condizioni il virus è più propenso a «remixare» i suoi geni e generare ceppi nuovi e potenzialmente più pericolosi — e come possiamo individuare in anticipo queste zone di rischio?
Seguire un virus che cambia forma
I virus influenzali trasportano il loro materiale genetico in otto frammenti separati, che possono essere scambiati quando due ceppi diversi infettano lo stesso uccello. Questo processo, chiamato riassortimento, può creare combinazioni virali completamente nuove. I ricercatori hanno raccolto più di 300.000 sequenze geniche influenzali da banche dati globali e, utilizzando una pipeline standardizzata, le hanno raggruppate in famiglie genetiche per ciascuno degli otto segmenti. Hanno quindi definito 136 «genotipi» genetici distinti dei virus H5 altamente patogeni circolanti a livello mondiale tra il 1996 e il 2023. Tracciando dove e quando questi genotipi sono comparsi, hanno potuto ricostruire il panorama in evoluzione dei virus H5 nel tempo.
Tre ondate di cambiamento virale
Il team ha rilevato che l’evoluzione dell’H5 in Eurasia si è svolta in tre ampie ondate. Dal 2000 al 2013 un genotipo principale ha dominato gli focolai, soprattutto in Asia e in alcune parti dell’Africa, provocando eventi sporadici ma gravi negli allevamenti di pollame. Intorno al 2014 è emerso un nuovo ramo di H5, noto come clade 2.3.4.4, che ha inaugurato una seconda ondata. Tra il 2014 e il 2021 molti diversi genotipi sono coesistiti e si sono diffusi sia attraverso uccelli selvatici sia tramite allevamenti domestici, in particolare in Europa, Asia e successivamente nelle Americhe. Una terza ondata è iniziata intorno al 2021 con l’ascesa di clade 2.3.4.4b H5N1, che si è radicato in diverse regioni e ha causato focolai tutto l’anno — un andamento «endemico» piuttosto che picchi invernali occasionali. 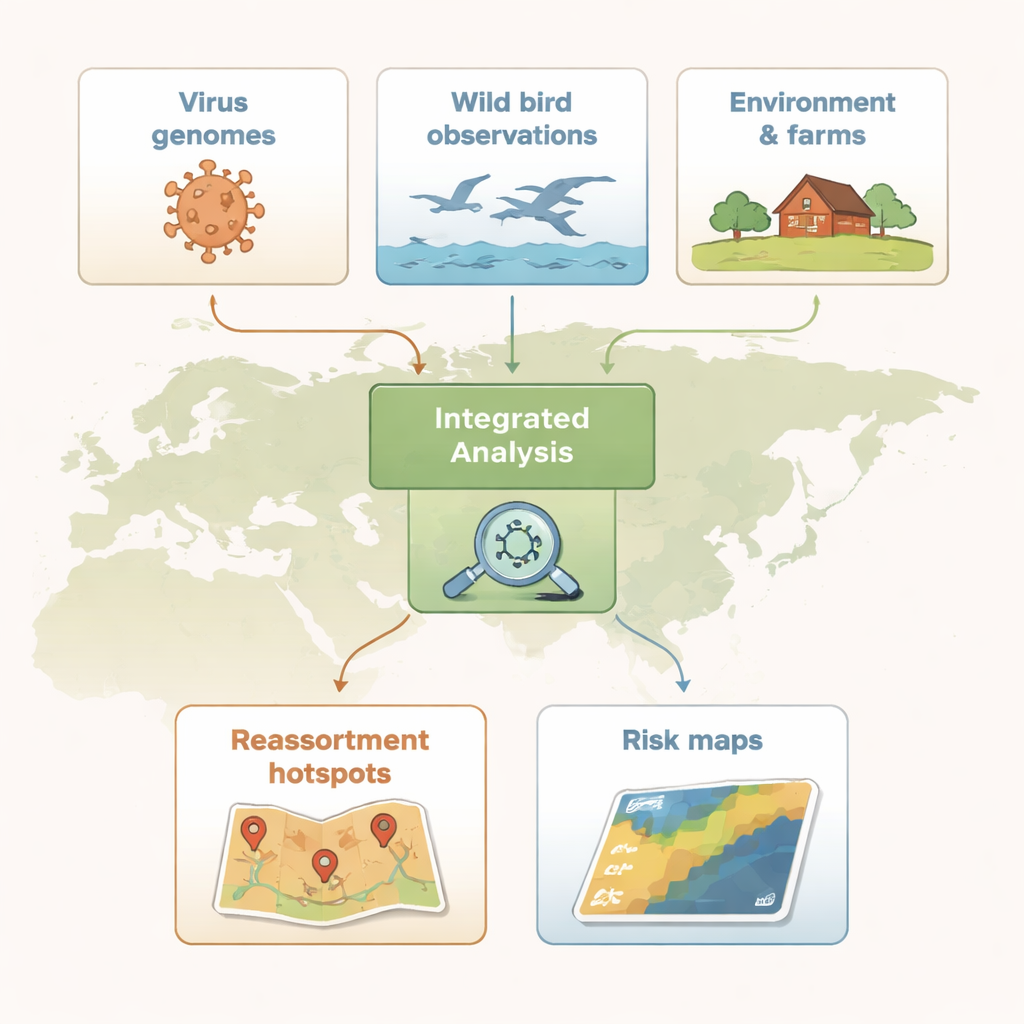
Cartografare hotspot nascosti
Per individuare dove lo scambio genico era più intenso, gli scienziati hanno suddiviso l’Eurasia in quadrati di griglia da 100 chilometri e hanno contato quanti diversi genotipi H5 sono stati rilevati in ciascuno. Utilizzando una statistica spaziale che mette in evidenza i cluster, hanno identificato hotspot di riassortimento — aree dove molti genotipi si presentavano insieme più spesso del previsto. All’inizio questi hotspot erano concentrati nel Sud-est asiatico. Nella seconda ondata si sono spostati verso nord e ovest, comparendo lungo le coste pacifiche dell’Asia orientale e attraverso l’Europa centrale e occidentale, incluse regioni della Danimarca, del sud della Svezia e del nord Italia. Questi schemi suggerivano che sia la geografia sia le pratiche agricole stessero indirizzando l’evoluzione del virus.
Comunità di uccelli, allevamenti e ambiente
Gli hotspot non nascono da una singola specie «cattiva» di uccelli o da un solo tipo di allevamento; si sviluppano dove si sovrappongono molti fattori. Il team ha combinato segnalazioni di avvistamenti di uccelli di citizen science dal progetto eBird con mappe di uso del suolo, dati sulla densità del pollame e registri di focolai di H5 negli allevamenti. Hanno prima identificato le specie di uccelli selvatici che tendevano a essere presenti nelle griglie hotspot, concentrandosi su tre principali ordini di uccelli: anatidi come anatre e oche (Anseriformes), limicoli (Charadriiformes) e passeriformi (Passeriformes). Sorprendentemente, molte specie ad alto rischio non erano mai state testate formalmente per l’influenza aviaria. Per catturare l’effetto combinato di più specie, gli autori hanno costruito un «punteggio di rischio polispecie» che sintetizza quanto la comunità di uccelli di una località fosse in grado di favorire il riassortimento. Hanno poi aggiunto informazioni su densità di polli e anatre, focolai negli allevamenti e tipi di territorio come aree coltivate o urbane per stimare quali combinazioni di condizioni predicevano più fortemente gli hotspot.
Dalle zone umide ai capannoni di pollame
L’analisi ha rivelato uno spostamento nella nicchia ecologica del virus. Nei primi anni il riassortimento era legato principalmente all’allevamento di anatre, coerente con il ruolo delle anatre come serbatoio silenzioso che ospita il virus senza sintomi evidenti. Con il tempo, man mano che i virus H5 altamente patogeni si sono stabilizzati negli allevamenti di polli — favorito in alcune regioni da circolazione prolungata e pratiche vaccinali — i segnali più forti si sono spostati verso aree ad alta densità di polli e paesaggi agricoli misti. Aree urbanizzate in parti dell’Asia e terre coltivate in Europa hanno inoltre mostrato correlazioni con gli hotspot, probabilmente riflettendo i punti di incontro tra persone, allevamenti e uccelli selvatici. Allo stesso tempo, uccelli non acquatici come i passeriformi, che vivono in grandi numeri attorno a campi, sobborghi e fienili, sono apparsi sempre più come un ponte tra habitat selvatici e capannoni di pollame. 
Cosa significa per la preparazione
Per i non specialisti, il messaggio essenziale è che nuove forme pericolose di influenza aviaria H5 sono più probabili dove si incontrano allevamenti intensivi di pollame, comunità avifaunistiche diversificate e paesaggi modificati dall’uomo. Integrando dati genetici, registri di osservazione degli uccelli e informazioni ambientali in mappe di rischio unificate, questo studio offre una guida su dove la sorveglianza può essere più efficace — che si tratti di testare gruppi di uccelli poco studiati, rafforzare la biosicurezza attorno agli allevamenti ad alto rischio o monitorare regioni dove il virus è diventato endemico. Capire e sorvegliare queste «zone di remix» genetico è un passo pratico per ridurre la probabilità che un virus animale ci sorprenda con un’ulteriore salto in espansione, virulenza o specie ospite.
Citazione: Chen, BJ., Liang, CC., Li, YT. et al. Integrated approaches to explore temporal-spatial changes in gene reassortment of highly pathogenic avian influenza A(H5) virus in Eurasia, 2000–2023. Sci Rep 16, 7518 (2026). https://doi.org/10.1038/s41598-026-38466-y
Parole chiave: influenza aviaria, H5N1, uccelli selvatici, allevamento di pollame, evoluzione virale