Clear Sky Science · it
Caratterizzazione molecolare dei disturbi atassici e neuropatici a trasmissione recessiva in famiglie pakistane consanguinee
Perché è importante per le famiglie e i medici
I problemi di equilibrio, deambulazione e sensibilità nelle mani e nei piedi possono essere profondamente invalidanti, soprattutto quando insorgono in età infantile e peggiorano lentamente nel corso della vita. Per molte famiglie, in particolare in regioni dove i matrimoni tra cugini sono frequenti, questi sintomi si ripetono per più generazioni senza una spiegazione chiara. Questo studio affronta una questione urgente per tali famiglie in Pakistan: l’analisi moderna del DNA può finalmente rivelare le cause genetiche nascoste della loro atassia (disturbi dell’equilibrio e della coordinazione) e neuropatia periferica (danno ai nervi degli arti), e aiutare i medici a offrire diagnosi più precise e potenziali terapie?
Seguire la malattia ereditata attraverso famiglie numerose
I ricercatori hanno lavorato con sette famiglie pakistane estese in cui diversi membri presentavano gravi disturbi del movimento e dei nervi. Alcune persone avevano predominantemente atassia, che rende difficile camminare con stabilità o controllare il linguaggio e i movimenti oculari. Altri mostravano segni classici di neuropatia periferica, come perdita di massa muscolare in mani e piedi, deformità plantari e perdita dei riflessi. In queste famiglie i genitori erano imparentati tra loro, aumentando la probabilità che i figli ereditassero due copie dello stesso gene raro difettoso. Usando campioni di sangue di parenti affetti e non affetti, il team ha eseguito il sequenziamento dell’esoma—lettura di quasi tutte le porzioni codificanti proteine del genoma—per cercare variazioni dannose che seguissero la malattia attraverso gli alberi genealogici. 
Individuare geni rari difettosi
Filtrando le differenze del DNA comuni e benigne, gli scienziati hanno identificato varianti probabilmente responsabili della malattia in cinque delle sette famiglie. Ciascuna di queste famiglie portava una specifica modifica genetica, e tutte seguivano un modello recessivo: la malattia si manifestava solo quando si ereditavano due copie difettose, una da ciascun genitore. In una famiglia con problemi di equilibrio insorti in età adulta e difficoltà del linguaggio, il colpevole è stata una variante rara nel gene MFSD8, che contribuisce al trasporto di materiali nei compartimenti di riciclo cellulare chiamati lisosomi. In un’altra famiglia, una variante dannosa in AFG3L2, una proteina che mantiene la salute dei mitocondri—le centrali energetiche della cellula—è stata collegata a un’atassia spastica con distonia ad esordio infantile (contrazioni muscolari anomale). Una terza famiglia portava un errore frameshift in SETX, un gene che protegge il DNA durante la riparazione ed è già noto per causare atassia con apraxia oculomotoria, un disturbo che colpisce anche i movimenti oculari.
Uno sguardo più attento al danno nervoso ereditario
Due ulteriori famiglie presentavano una forma della malattia di Charcot‑Marie‑Tooth (CMT), un gruppo di condizioni ereditarie che danneggiano i nervi lunghi diretto verso piedi e mani. In entrambe, i ricercatori hanno trovato varianti dannose in un gene chiamato GDAP1, cruciale per la normale funzione mitocondriale nelle cellule nervose. Una variante di GDAP1 tronca la proteina ed è stata associata a una forma molto grave con esordio precoce; un’altra sostituisce un singolo aminoacido nella proteina e determina un decorso relativamente più lieve. Colpisce che il paziente più gravemente colpito in una delle famiglie CMT fosse anche omozigote per una variante nota di malattia in un secondo gene, MMACHC, coinvolto nel metabolismo della vitamina B12 e talvolta trattabile con terapie a base di vitamine. Questo doppio colpo potrebbe spiegare perché i suoi sintomi fossero più severi rispetto a quelli dei parenti che non avevano la variante MMACHC.
Quando la ricerca del DNA non basta
Non tutte le famiglie hanno fornito una risposta genetica chiara. In due delle sette famiglie, il team non è riuscito a trovare alcuna singola variante nell’esoma che corrispondesse in modo convincente al pattern di malattia. In un caso hanno identificato una variante nel gene EHHADH che seguiva il modello di ereditarietà ma è prevista come benigna ed è nota per causare una diversa patologia renale quando alterata. In un altro, due cugini con problemi di movimento simili si sono rivelati avere cause sottostanti differenti: un ragazzo portava una variante nota dannosa in ALS2, che può portare a forme giovanili di malattia del motoneurone, mentre i suoi cugini affetti non la possedevano. Questi casi non risolti suggeriscono che mutazioni importanti possano trovarsi in regioni del genoma non catturate dal sequenziamento dell’esoma standard, o che più fattori genetici sottili possano interagire tra loro.
Cosa significa per i pazienti e la cura futura
Nel complesso, i risultati mostrano che potenti strumenti genetici possono scoprire i geni specifici alla base di complessi disturbi del sistema nervoso e dell’equilibrio, anche in contesti con risorse limitate. Per le cinque famiglie con risultati chiari, il lavoro trasforma etichette vaghe come “atassia” o “neuropatia” in diagnosi precise legate a determinati geni, utili per la consulenza genetica, per delineare la prognosi e, in alcuni casi, per evidenziare opzioni terapeutiche — come le terapie collegate alla vitamina B12 per le malattie associate a MMACHC. Lo studio amplia anche la comprensione di come geni come MFSD8, AFG3L2, SETX, GDAP1, MMACHC e ALS2 influenzino la salute delle cellule nervose nel cervello, nel midollo spinale e nei nervi periferici. Guardando avanti, saranno necessari sequenziamenti genomici più completi e studi funzionali per risolvere i misteri rimanenti e tradurre queste informazioni genetiche in una diagnosi più precoce e in cure migliori per i bambini e gli adulti colpiti. 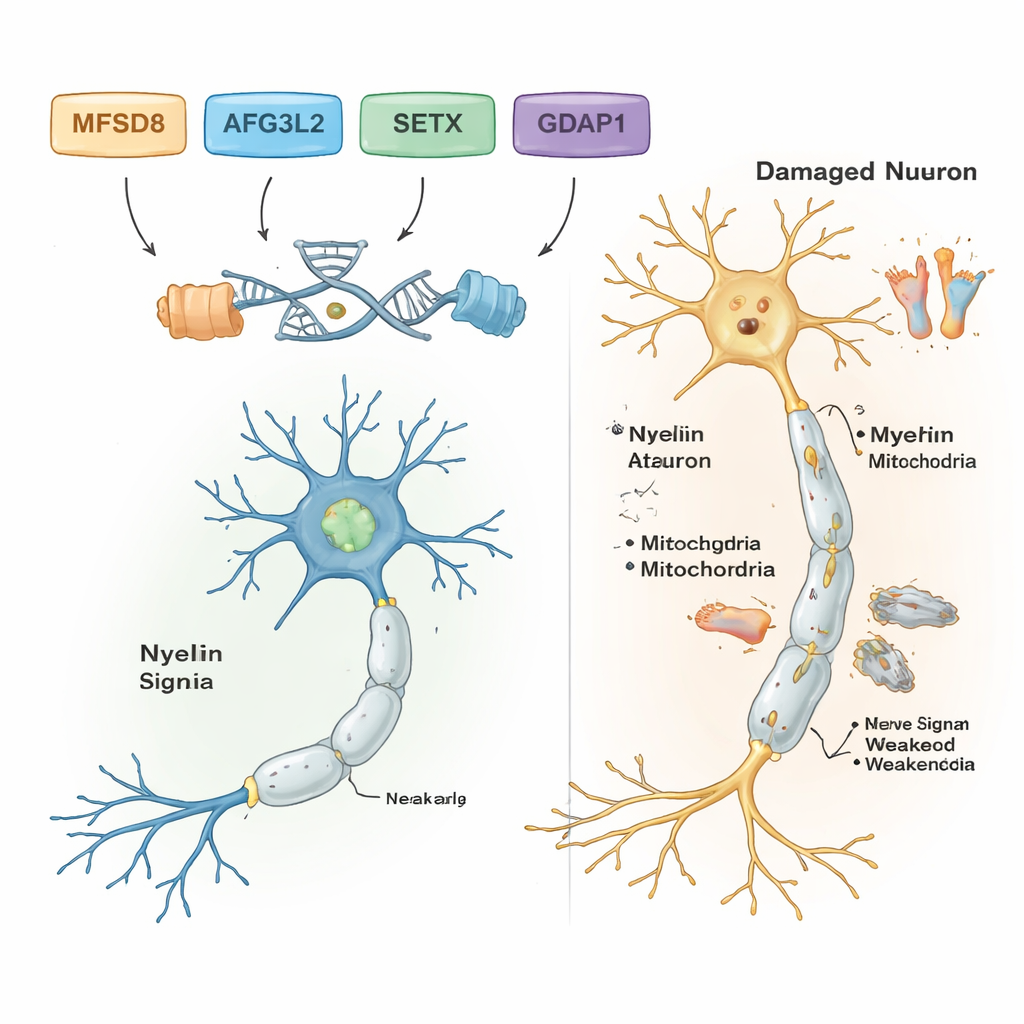
Citazione: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Parole chiave: atassia, neuropatia periferica, sequenziamento dell'esoma, malattia di Charcot-Marie-Tooth, diagnosi genetica