Clear Sky Science · it
Ottimizzazione della selezione delle caratteristiche nei dati di microarray per il cancro usando un framework evolutivo guidato da heap per spazi ad alta dimensionalità
Perché scegliere i geni giusti è importante
I test per il cancro basati sulle moderne tecnologie genetiche possono misurare decine di migliaia di geni contemporaneamente, ma i medici spesso dispongono di dati provenienti da solo poche dozzine di pazienti. Nella vasta “giungla genica” si nasconde un numero molto più piccolo di segnali che distinguono realmente un tipo di cancro da un altro, o un tumore da tessuto sano. Questo articolo presenta un nuovo metodo di ricerca intelligente per selezionare automaticamente quei geni chiave, con l’obiettivo di rendere la diagnosi assistita dal computer più accurata, più veloce e più facile da interpretare.
Troppi segnali, pochi dati
Gli esperimenti su microarray e tecnologie affini consentono ai ricercatori di misurare i livelli di attività di migliaia di geni in ciascun campione di paziente. Tuttavia il numero di campioni è di solito molto basso, a volte inferiore a cento. Molte di queste letture geniche sono rumorose, ridondanti o irrilevanti per la malattia in esame. Conservare tutte le misure può sovraccaricare gli algoritmi di apprendimento, rallentare i calcoli e produrre modelli fuorvianti che si attaccano a casualità anziché alla vera biologia. Il processo di riduzione a un sottoinsieme utile si chiama “selezione delle caratteristiche” ed è cruciale se vogliamo previsioni affidabili dai dati medici ad alta dimensionalità.
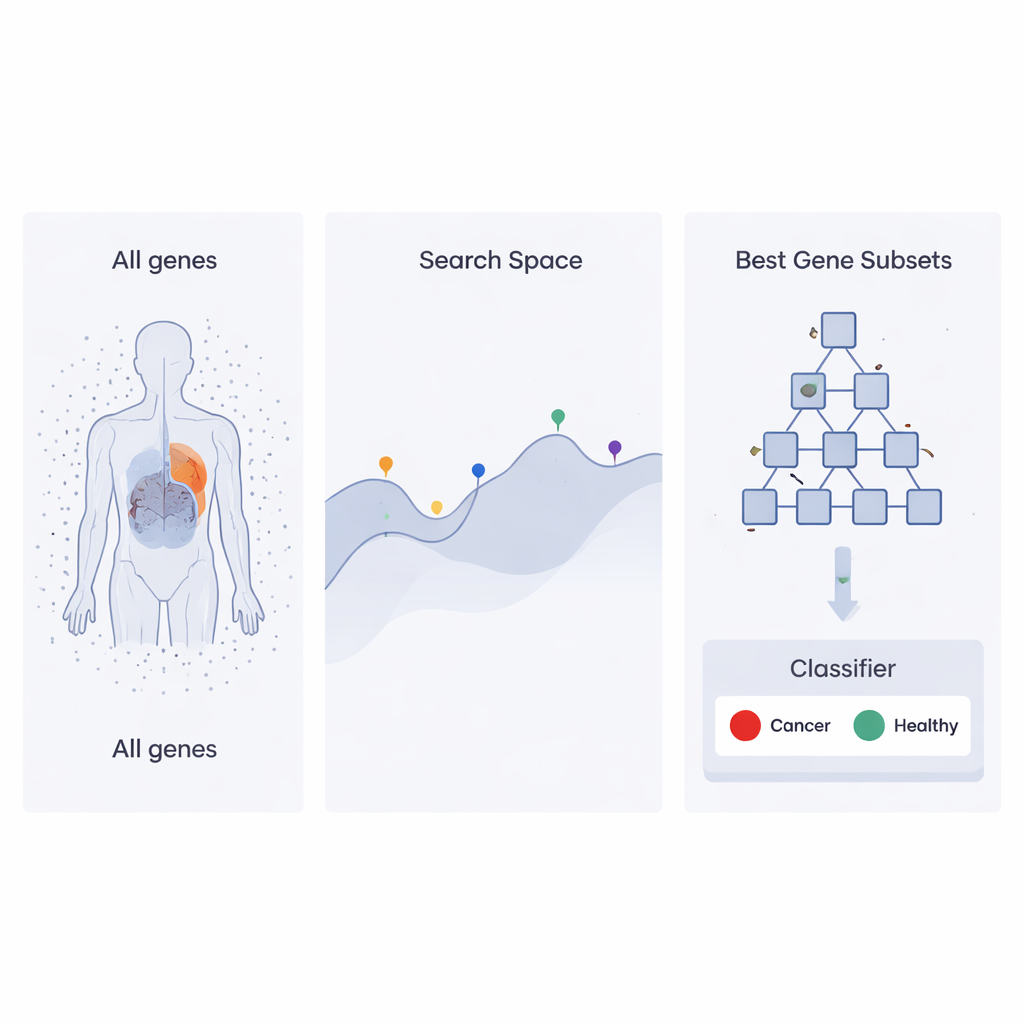
Una strategia di ricerca ispirata alle gerarchie aziendali
Gli autori si basano su un recente approccio di ottimizzazione chiamato Heap‑Based Optimizer (HBO), che prende spunto dall’organizzazione dei dipendenti in un’azienda. Immaginate ogni possibile insieme di geni come un “dipendente” la cui prestazione lavorativa è giudicata da quanto bene aiuta un classificatore a distinguere i campioni tumorali da quelli sani. Questi dipendenti sono disposti in una gerarchia, come una scala aziendale, usando una struttura dati nota come heap. Gli insiemi di geni con prestazioni elevate si posizionano vicino alla cima, mentre quelli più deboli stanno più in basso. Nel corso di molte iterazioni, i dipendenti di rango inferiore adeguano le loro scelte copiando e modificando leggermente ciò che fanno i loro capi e colleghi, spingendo gradualmente l’intera organizzazione verso soluzioni migliori.
Trasformare i dati grezzi in pattern più netti
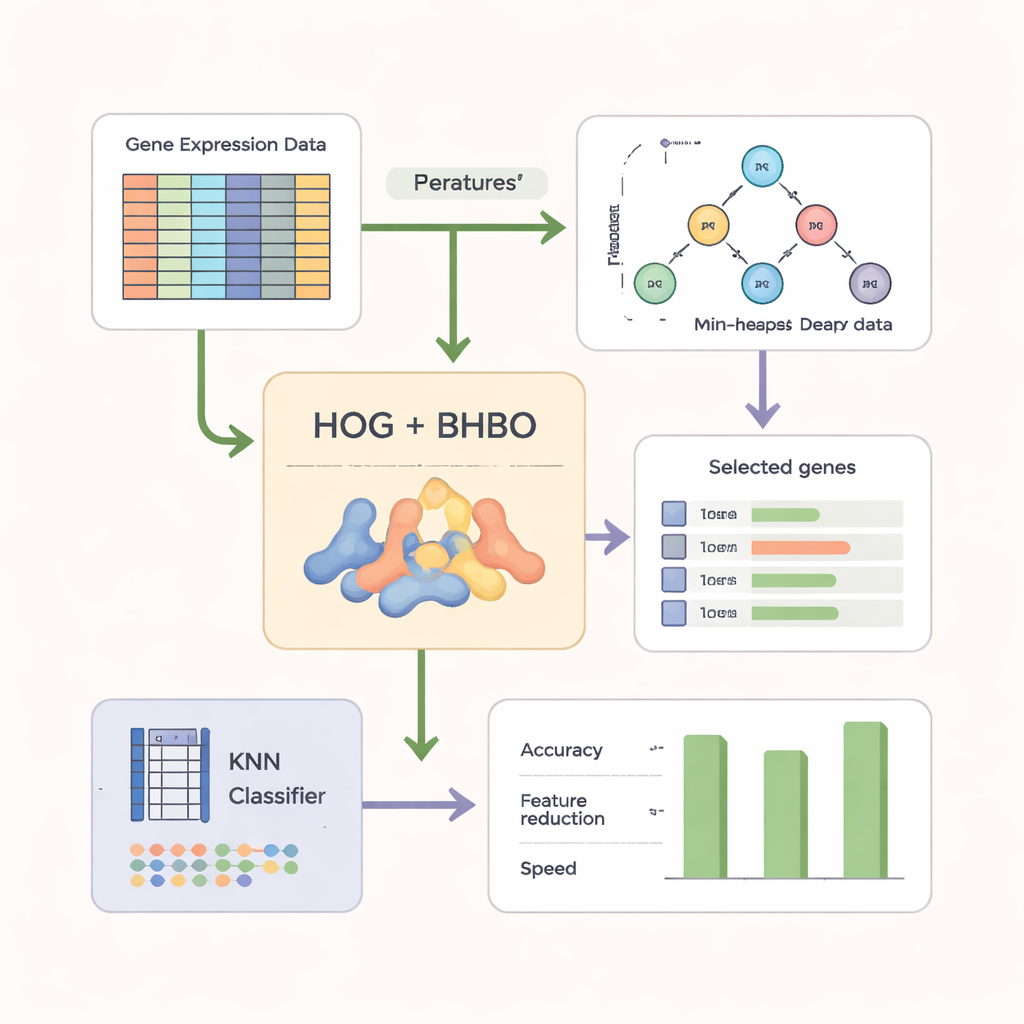
Per rendere la ricerca più efficace, gli autori non si affidano solo alle letture geniche grezze. Prima ristrutturano i dati di microarray in una forma simile a un’immagine e applicano una tecnica chiamata Histogram of Oriented Gradients (HOG), ampiamente usata nella visione artificiale. HOG cattura come cambiano i livelli di espressione attraverso i geni, mettendo in evidenza pattern locali anziché misurazioni isolate. Queste caratteristiche basate sui pattern vengono quindi combinate con le informazioni geniche originali. Un classificatore semplice chiamato k‑Nearest Neighbors (KNN) funge da “giudice”, valutando ogni sottoinsieme di geni candidato in base a quanto accuratamente etichetta nuovi campioni e premiando anche insiemi più piccoli e compatti.
Test su più dataset di cancro
I ricercatori hanno valutato la loro versione binaria del Heap‑Based Optimizer (BHBO) su nove dataset pubblici di microarray per il cancro, inclusi tumori cerebrali, leucemie, cancro alla prostata e collezioni miste di tumori con molti sottotipi. Ogni dataset conteneva da migliaia a oltre quindicimila geni misurati ma relativamente pochi campioni di pazienti. Per ciascun dataset, BHBO è stato eseguito ripetutamente e confrontato con sette metodi di ricerca noti, come algoritmi genetici e ottimizzazione a sciame di particelle. Il team ha misurato non solo l’accuratezza, ma anche quante caratteristiche sono state mantenute, la rapidità di convergenza della ricerca e la stabilità dei risultati quando i dati venivano disturbati con rumore simulato, effetti batch e errori di etichettatura.
Cosa ha ottenuto il nuovo metodo
Nei nove dataset, l’approccio guidato dall’heap ha raggiunto in media un’accuratezza di classificazione di circa il 95 percento riducendo il numero di geni di oltre l’85 percento. Ha chiaramente battuto i metodi concorrenti in diversi dataset e ha mostrato una convergenza più rapida, cioè ha individuato buoni insiemi di geni in meno passi di ricerca. Anche quando gli autori hanno deliberatamente corrotto i dati — aggiungendo rumore o invertendo alcune etichette dei campioni — le prestazioni del metodo sono calate solo leggermente e sono rimaste superiori alle alternative. Test statistici hanno confermato che questi miglioramenti sono improbabili dovuti al caso.
Cosa significa per il futuro della diagnostica oncologica
In termini pratici, questo lavoro dimostra che una strategia di ricerca progettata con cura può setacciare enormi dataset genetici e scoprire piccoli pannelli di geni ricchi di informazione che classificano comunque molto bene i tumori. Per clinici e ricercatori, insiemi genici così compatti sono più facili da validare biologicamente, meno costosi da misurare in test di follow‑up e più adatti all’integrazione in strumenti di supporto alle decisioni. Pur non scoprendo direttamente nuovi farmaci o percorsi biologici, il metodo concentra l’attenzione su marcatori genetici promettenti, aiutando altri studi a focalizzarsi sui segnali più informativi nascosti nei dati oncologici ad alta dimensionalità.
Citazione: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Parole chiave: microarray per il cancro, selezione delle caratteristiche, ottimizzazione metaeuristica, biomarcatori genetici, data mining medico