Clear Sky Science · it
Un metodo per il rilevamento delle varianti strutturali usando la matrice di contatti Hi-C e reti neurali
Perché la curvatura del DNA in 3D è importante
Il nostro DNA viene spesso rappresentato come una semplice stringa di lettere, ma all'interno di ciascuna cellula si ripiega in una forma tridimensionale complessa. Quando grandi porzioni di questa stringa vengono eliminate, capovolte o spostate — cambiamenti detti variazioni strutturali — possono interrompere geni e contribuire allo sviluppo del cancro. Questo studio presenta VarHiCNet, un nuovo sistema di intelligenza artificiale che interpreta le mappe di ripiegamento 3D del DNA e individua questi cambiamenti su larga scala con maggiore precisione rispetto agli strumenti esistenti, offrendo un nuovo approccio per studiare i genomi tumorali e altre malattie.
Vedere i cambiamenti del genoma tramite mappe di contatto 3D
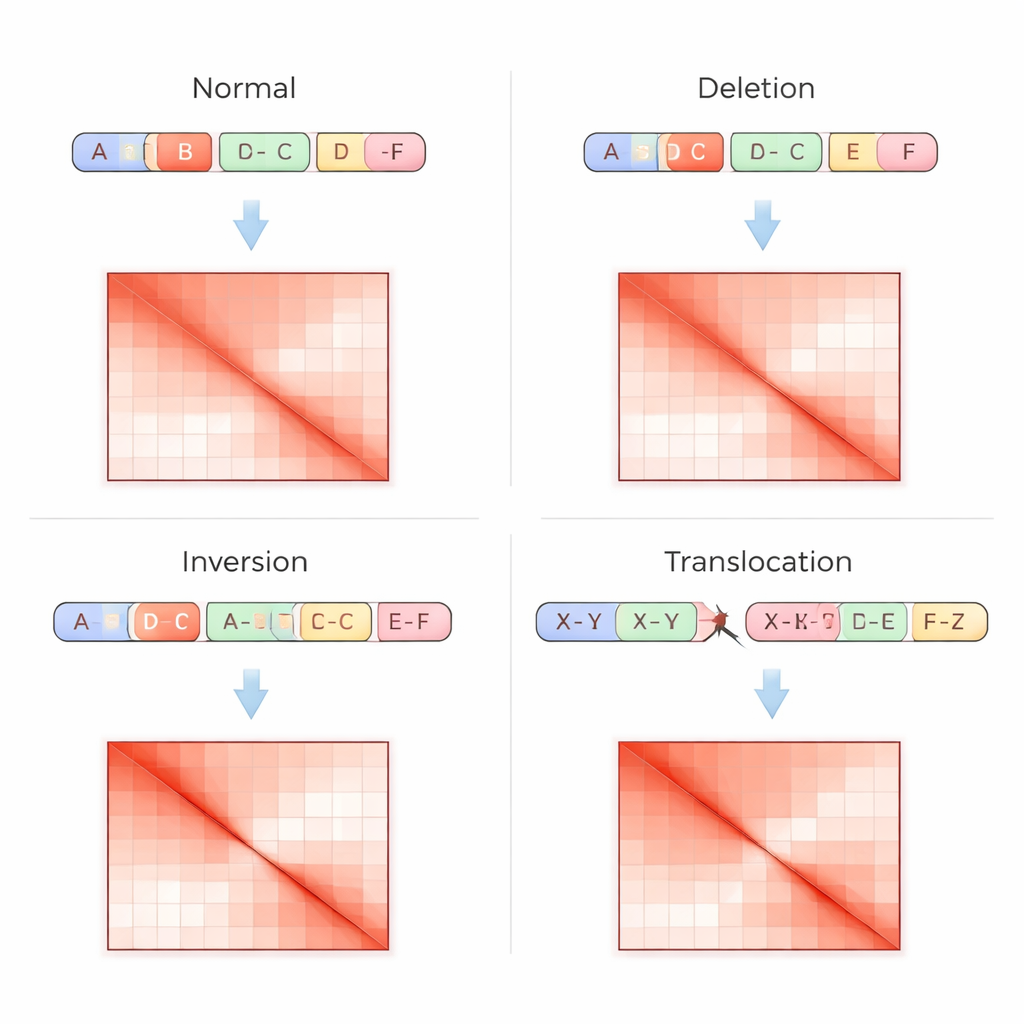
I test genomici tradizionali leggono il DNA come una sequenza lineare, il che rende difficile rilevare riorganizzazioni complesse, soprattutto nelle regioni ripetitive o quando i segmenti si spostano senza cambiare il numero di copie. La tecnica Hi-C affronta il problema in modo diverso: misura quanto spesso parti distanti del DNA entrano in contatto fisico all'interno del nucleo e registra questi contatti come una griglia, o matrice di contatto, dove i punti più luminosi indicano interazioni più forti. Le variazioni strutturali lasciano impronte distintive in queste matrici — come bande mancanti dove una regione è stata cancellata, schemi specchiati quando un segmento è invertito, o hotspot fuori diagonale dove due cromosomi si sono fusi. VarHiCNet è progettato per riconoscere automaticamente questi pattern visivi.
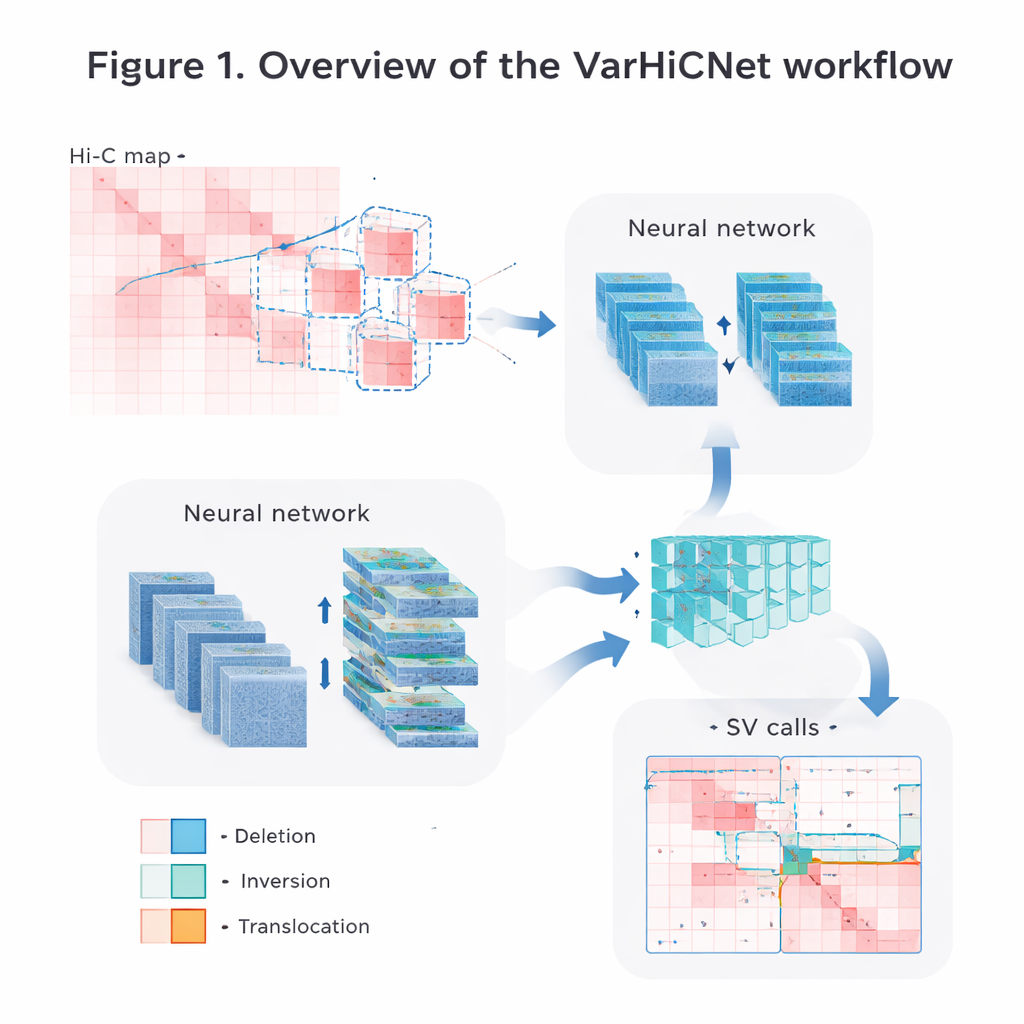
Trasformare le mappe del genoma in immagini per l'IA
Gli autori convertono i dati grezzi dei contatti Hi-C in immagini che i sistemi di visione artificiale possono elaborare facilmente. Innanzitutto normalizzano con cura le matrici per correggere il naturale calo di frequenza dei contatti al crescere della distanza tra i segmenti di DNA, preservando al contempo sia i segnali di interazione ravvicinata sia quelli a lungo raggio. Poi scansionano ogni cromosoma con finestre quadrate sovrapposte e ritagliano molte sotto-matrici più piccole. Ogni sotto-matrice viene ridimensionata in un'immagine a colori standardizzata di 800 per 800 pixel, dove diverse intensità di contatto sono mappate in tonalità di rosso su tre canali colore. Questa rappresentazione simil-immagine permette al modello di riutilizzare potenti tecniche originariamente sviluppate per il riconoscimento di oggetti nelle fotografie.
Prendere in prestito trucchi dal rilevamento di oggetti
VarHiCNet tratta ogni potenziale variante strutturale come se fosse un “oggetto” in un'immagine. Si basa su un moderno framework di object detection chiamato RT-DETR, che utilizza una combinazione di reti convoluzionali e Transformer per evidenziare le regioni importanti. Un backbone ResNet estrae prima caratteristiche multi-scala: gli strati superficiali mantengono dettagli fini necessari per individuare con precisione i punti di rottura, mentre gli strati più profondi catturano pattern più ampi che indicano eventi su vasta scala. Un modulo di fusione delle caratteristiche poi combina informazioni provenienti da diversi strati in modo che siano preservati sia gli indizi locali sia quelli globali. Un altro blocco personalizzato, ispirato allo spatial pyramid pooling, regola quanto della regione circostante il modello “vede” contemporaneamente, rendendolo sensibile a varianti che coprono tratti di DNA sia relativamente piccoli sia molto estesi.
Dalle regioni candidate ai tipi di variante precisi
Una volta che VarHiCNet ha proposto regioni candidate nell'immagine Hi-C, deve affinarle in punti di rottura esatti e in tipi di variante specifici, come delezioni, inversioni, duplicazioni o traslocazioni. Per farlo, il sistema ingrandisce il vicinato attorno a ciascun punto di rottura previsto e ne riduce la complessità usando una tecnica matematica chiamata analisi delle componenti principali (PCA), che mette in evidenza dove il pattern di contatto cambia più bruscamente. Queste rappresentazioni compatte vengono quindi fornite a un classificatore basato su Transformer che apprende le sottili differenze nei pattern locali per ciascuna categoria di variante. Il risultato è una chiamata dettagliata per ogni evento: dove avviene nel genoma e che tipo di cambiamento strutturale rappresenta.
Prestazioni su diverse linee cellulari tumorali
I ricercatori hanno testato VarHiCNet su dati Hi-C di sei diverse linee cellulari tumorali umane, coprendo tumori del sangue, seno, cervello, rene, polmone e prostata. Usando un catalogo ad alta confidenza di varianti strutturali note come standard di riferimento, hanno confrontato il loro metodo con diversi strumenti di punta che analizzano anch'essi i dati Hi-C. Sia per eventi all'interno dello stesso cromosoma sia per eventi tra cromosomi, VarHiCNet ha generalmente raggiunto punteggi F1 maggiori o comparabili, il che significa che bilanciava meglio sensibilità e accuratezza rispetto ad altri approcci. È risultato particolarmente efficace nel rilevare traslocazioni bilanciate e inversioni — riorganizzazioni che spesso lasciano poche tracce nel sequenziamento tradizionale ma che lasciano chiare firme nel ripiegamento 3D. Gli autori hanno inoltre dimostrato che le loro scelte progettuali, come la risoluzione delle immagini e i moduli di fusione delle caratteristiche, miglioravano costantemente le prestazioni nei test controllati.
Cosa significa per la comprensione delle malattie
In termini pratici, VarHiCNet offre ai ricercatori un modo più intelligente per “guardare” come il genoma si ripiega in 3D e per individuare grandi riorganizzazioni legate a malattie che potrebbero sfuggire al sequenziamento convenzionale. Trasformando mappe di contatto complesse in immagini e applicando moderne reti neurali di tipo visivo, il metodo può rilevare e classificare molti tipi di variazioni strutturali con alta affidabilità attraverso diversi tipi di cellule tumorali. Pur rimanendo limitato su alcune varianti molto piccole o altamente complesse e dipendendo da dati di addestramento ricchi, VarHiCNet indica un futuro in cui l'architettura 3D del genoma diventerà una parte routine di come leggiamo, interpretiamo e, in ultima analisi, indirizziamo i cambiamenti genetici alla base del cancro e di altre malattie.
Citazione: Shen, J., Wang, H., Zhai, H. et al. A method for structural variant detection using Hi-C contact matrix and neural networks. Sci Rep 16, 7324 (2026). https://doi.org/10.1038/s41598-026-37678-6
Parole chiave: variazione strutturale, Hi-C, deep learning, genomica del cancro, genoma 3D