Clear Sky Science · it
Caratterizzazione in vitro del dominio catalitico della deacetilasi delle istoni umana 5
Perché piccoli interruttori nell’impacchettamento del DNA sono importanti
All’interno di ogni cellula il nostro DNA è avvolto attorno a proteine che fungono da rocchetti, permettendo di far entrare metri di materiale genetico in uno spazio microscopico. L’attivazione o la repressione di un gene dipende spesso da piccole etichette chimiche su queste proteine-rocchetto. Questo studio si concentra su uno specifico “interruttore” proteico chiamato HDAC5, collegato a malattie cardiache, disturbi neurologici, cancro e altro. Comprendere il funzionamento molecolare di HDAC5 può aprire la strada a farmaci più mirati e con meno effetti collaterali.
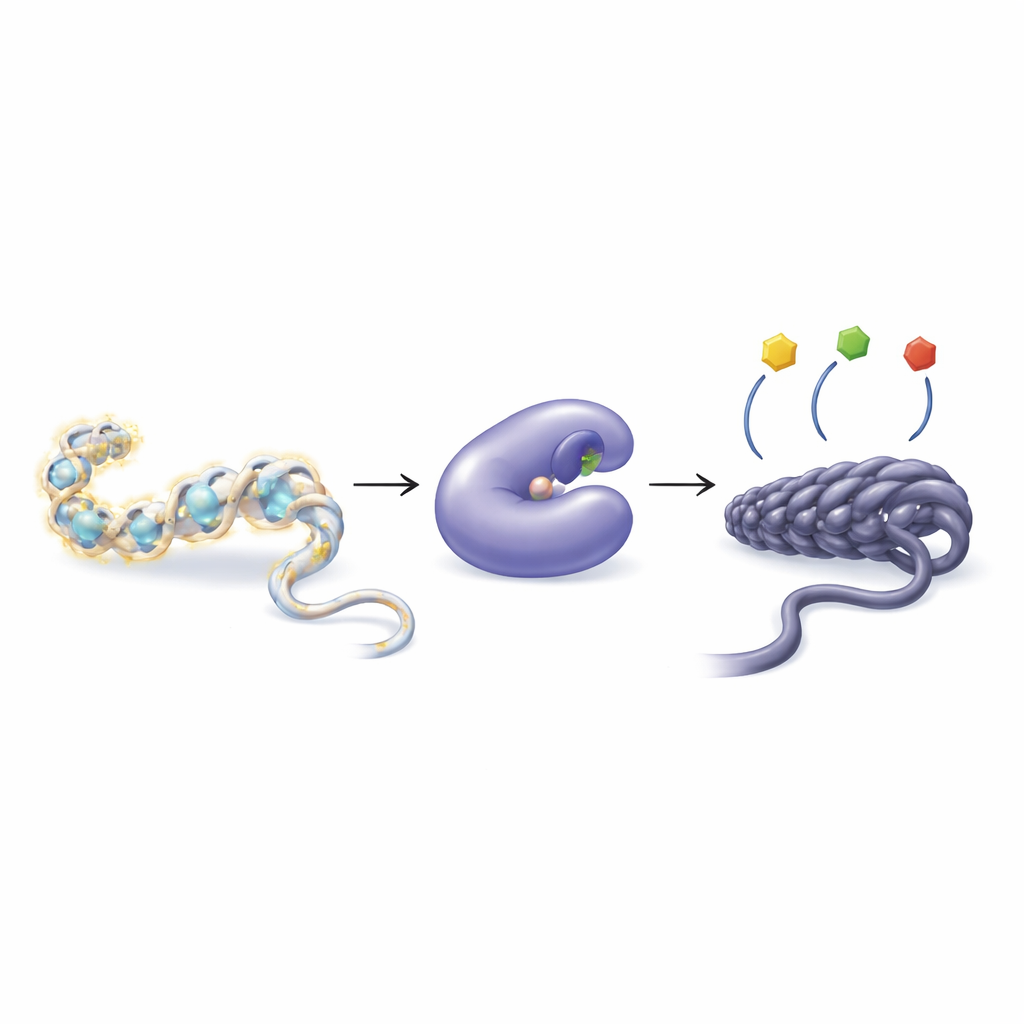
Come le cellule modulano i geni con piccole etichette chimiche
Il nostro DNA non galleggia liberamente ma è avvolto attorno a proteine chiamate istoni, formando una struttura nota come cromatina. Le cellule possono aggiungere o rimuovere piccoli gruppi chimici, come gruppi acetile, dalle code degli istoni per rendere la cromatina più sciolta o più compatta. Un impacchettamento più lasso facilita in genere la lettura dei geni; un impacchettamento più stretto tende a silenziarli. Due gruppi di enzimi gestiscono questo equilibrio: le acetiltransferasi degli istoni aggiungono gruppi acetile, mentre le deacetilasi degli istoni (HDAC) li rimuovono. Quando questo equilibrio viene alterato, può contribuire a una vasta gamma di malattie tra cui cancro, patologie cardiache, atrofia muscolare e disturbi del sistema immunitario.
Perché HDAC5 è un bersaglio promettente ma complesso
Le HDAC costituiscono una grande famiglia di enzimi correlati divisa in diverse classi. Molti dei farmaci attualmente in uso bloccano contemporaneamente diversi tipi di HDAC, il che può compromettere funzioni normali importanti e causare effetti collaterali intensi. Le HDAC di classe IIa, incluso HDAC5, si distinguono perché sono più abbondanti in tessuti specifici come cervello, cuore e muscolo scheletrico e collaborano con altre proteine per regolare reti geniche chiave. HDAC5 spesso funge da ponte, portando un partner altamente attivo (HDAC3) su geni specifici in modo che la cromatina venga compattata e quei geni silenziati. Grazie a questi ruoli mirati, HDAC5 è considerato un bersaglio interessante per farmaci più selettivi, ma è mancata finora una dettagliata caratterizzazione biochimica e una struttura ad alta risoluzione del suo nucleo attivo, rendendo difficile il progetto razionale di inibitori.
Ricostruire HDAC5 in provetta
Per colmare questa lacuna, i ricercatori hanno prodotto solo il nucleo catalitico umano di HDAC5 — la parte che esegue la reazione chimica — in batteri, lo hanno purificato e hanno confermato che forma una proteina stabile e monomerica in soluzione. Hanno poi testato la sua attività a diversi livelli di sale e pH. L’attività di HDAC5 è risultata robusta su un’ampia gamma di concentrazioni saline e ha mostrato un picco in condizioni leggermente basiche, simili a quelle interne a molte cellule. Usando speciali substrati fluorescenti, hanno scoperto che la forma naturale di HDAC5 riconosce solo un particolare tipo di substrato comunemente usato per sondare le HDAC di classe IIa. Ispirandosi a studi precedenti su HDAC correlate, hanno sostituito un singolo aminoacido (istidina) con una tirosina in una posizione critica. Sorprendentemente, questo piccolo cambiamento ha permesso alla versione mutante di processare efficacemente entrambi i tipi di substrati di prova, rivelando come una singola residuo nel sito attivo indirizzi le preferenze chimiche dell’enzima.
Testare e confrontare due nuovi candidati farmaci
Il gruppo ha poi esaminato due molecole sperimentali che bloccano HDAC5, note come NT160 e FFK24. Questi composti impiegano un nuovo gruppo chelante dello zinco che evita parte della tossicità e della scarsa selettività osservate con i vecchi farmaci a base di idrossamato. Misurando quanto ciascun inibitore rallentava HDAC5 in reazioni controllate, gli autori hanno determinato costanti di inibizione estremamente basse nell’ordine dei nanomolari, il che significa che entrambi i composti si legano saldamente all’enzima. NT160 si è legato in modo consistente circa dieci volte più forte di FFK24. Per capire il motivo, i ricercatori hanno usato docking computazionale con una struttura del nucleo di HDAC5 predetta da AlphaFold. Entrambi gli inibitori condividono una regione “testa” che si infila profondamente nella tasca attiva e contatta lo ion metallico, ma la coda di NT160 forma ulteriori contatti stabilizzanti con specifici aminoacidi nella tasca. Queste interazioni addizionali spiegano probabilmente la sua maggiore potenza.
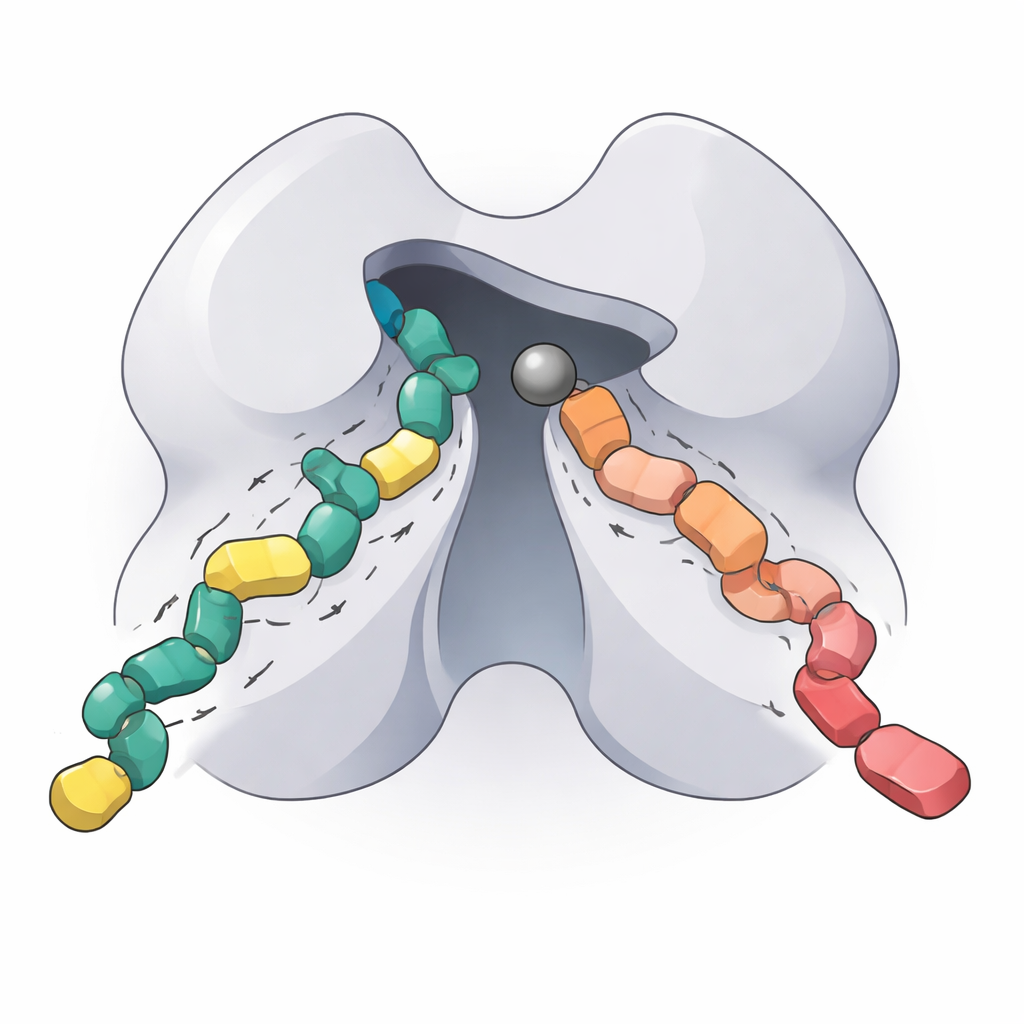
Cosa significa per le future terapie mirate
Ricostruendo il nucleo funzionale di HDAC5, mappandone le condizioni operative ottimali, spiegando come una singola sostituzione aminoacidica ne modifichi il comportamento e quantificando il legame di due inibitori di nuova generazione, questo studio fornisce un dettagliato “impronta” biochimica di un enzima importante ma precedentemente poco caratterizzato. Per i non specialisti, il messaggio chiave è che HDAC5 contribuisce a determinare se certi geni sono attivi o spenti e che modulare con precisione questo interruttore potrebbe essere utile nel trattamento di malattie cardiache, neurodegenerative, cancro e disturbi immunitari. Le nuove informazioni e gli strumenti presentati qui dovrebbero aiutare i ricercatori a progettare farmaci selettivi per HDAC5 e per le HDAC di classe IIa che agiscano dove necessario, riducendo al minimo effetti indesiderati in altre parti del corpo.
Citazione: Mammen, C., Hornung, F.M., Anzenhofer, C. et al. In vitro characterization of the catalytic domain of human histone deacetylase 5. Sci Rep 16, 7935 (2026). https://doi.org/10.1038/s41598-026-37633-5
Parole chiave: deacetilasi delle istoni 5, regolazione epigenetica, inibitori HDAC, terapia mirata contro il cancro, struttura della cromatina