Clear Sky Science · it
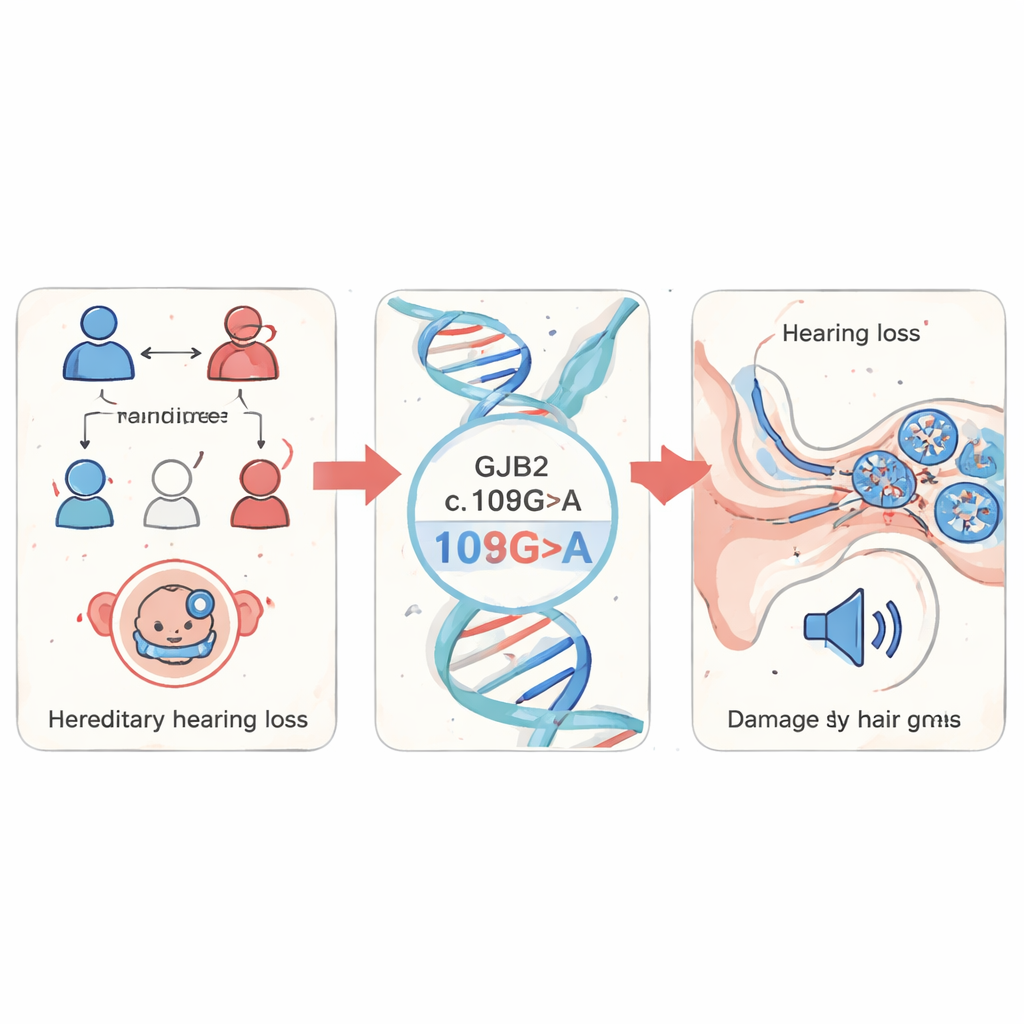
La mutazione GJB2 c.109G > A che attiva la via apoptotica mitocondriale mediata da IFI27 e porta a ipoacusia ereditaria non sindromica
Perché le minuscole cellule dell’orecchio contano per il futuro dei bambini
L’ipoacusia presente alla nascita interessa milioni di bambini nel mondo e spesso condiziona il modo in cui imparano a parlare, il successo scolastico e le relazioni sociali. Uno dei colpevoli genetici più comuni è il gene GJB2, ma i medici non avevano ancora compreso appieno come le variazioni in questo gene danneggino l’orecchio interno. Questo studio utilizza zebrafish e cellule umane per tracciare la catena di eventi da una singola modifica del DNA in GJB2 alla morte delle delicate cellule sensoriali dell’udito, indicando una nuova molecola, IFI27, come possibile bersaglio per futuri trattamenti.
Una mutazione comune dietro il silenzio infantile
I ricercatori hanno iniziato schermando campioni di sangue di 1.199 bambini con sospetta ipoacusia ereditaria nella provincia di Fujian, in Cina. Si sono concentrati su diversi geni della sordità ben noti e hanno riscontrato che le variazioni in GJB2 dominavano il quadro, rappresentando l’85% di tutte le mutazioni rilevate. Tra queste, una variante specifica chiamata c.109G>A (nota anche come p.Val37Ile) è risultata la più frequente. Questa variante è relativamente comune nella popolazione generale ma fortemente arricchita nelle persone con perdita uditiva, suggerendo un ruolo importante nell’ipoacusia non sindromica, cioè l’ipoacusia che si presenta senza altre patologie associate.
Seguire il danno in un pesce trasparente
Per osservare cosa fa questa mutazione in un organismo vivente, il gruppo ha usato lo zebrafish, un piccolo pesce d’acqua dolce i cui embrioni sono trasparenti e condividono molti geni e strutture auricolari con l’uomo. Hanno ingegnerizzato embrioni di zebrafish per esprimere sia la forma umana normale di GJB2 sia la versione mutata c.109G>A, e hanno inoltre impiegato un approccio di “knockdown” per ridurre l’espressione del gene gjb2 del pesce. Gli embrioni con il gene mutato o ridotto hanno mostrato crescita ritardata, code curvate e gonfiore attorno al cuore, segnali di sviluppo alterato. Soprattutto, le loro orecchie interne erano chiaramente anomale: strutture chiave chiamate otoliti risultavano più piccole e più distanti tra loro, e la regione cocleare piena di liquido era ridotta. Quando gli scienziati reintrodussero GJB2 normale insieme alla variante mutata, molti di questi difetti strutturali migliorarono, dimostrando che la mutazione stessa guidava le anomalie.
Dalle orecchie difettose a un comportamento uditivo alterato
Poiché l’udito dipende da minuscole “cellule ciliate” che convertono le vibrazioni sonore in segnali nervosi, il team ha colorato queste cellule negli zebrafish. I pesci con la mutazione GJB2 o con il knockdown presentavano molte meno cellule ciliate sia nell’orecchio interno sia lungo la superficie corporea, dove gli zebrafish percepiscono anche i movimenti dell’acqua. I ricercatori hanno poi testato la risposta dei pesci al suono. Utilizzando un sistema di tracciamento automatizzato, hanno misurato quanto lontano e quanto velocemente i larve di 5 giorni nuotassero quando esposti a brevi impulsi sonori. I pesci normali e quelli con GJB2 selvatico reagivano nuotando di più e più velocemente, mentre i pesci mutanti e con knockdown cambiavano a malapena il comportamento, indicando un deficit uditivo. Anche in questo caso, l’aggiunta di GJB2 normale ha in parte ripristinato sia il numero di cellule ciliate sia il movimento indotto dal suono.
Una via di morte all’interno delle centrali energetiche cellulari
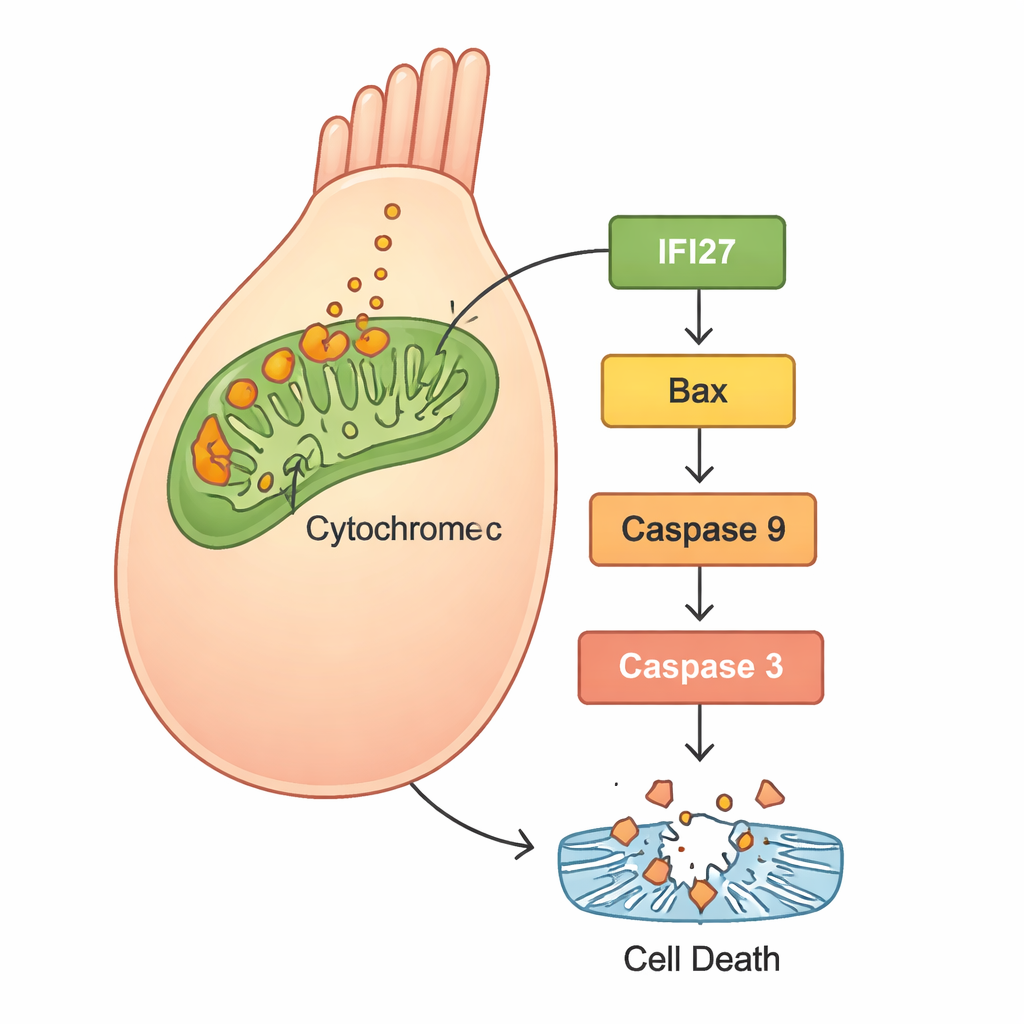
Per capire cosa accadeva all’interno delle cellule, gli scienziati hanno usato il sequenziamento dell’RNA per confrontare l’attività genica tra zebrafish normali e quelli con gjb2 ridotto. Un gruppo di geni collegati alla “via apoptotica mitocondriale” — una via di autodistruzione centrata nelle centrali energetiche della cellula — risultò fortemente attivato. In particolare emersero diversi membri della famiglia IFI27, insieme a noti attori della morte cellulare come Bax, citocromo c, Apaf1 e caspasi. Esperimenti successivi in cellule umane HEK293 confermarono il quadro: le cellule con GJB2 mutato producevano più specie reattive dell’ossigeno (ROS, una forma di stress ossidativo), rilasciavano più citocromo c dai mitocondri e attivavano proteine apoptotiche, portando a un aumento della morte cellulare. Quando i ricercatori silenziarono IFI27 in cellule portatrici del gene mutato, i livelli di ROS diminuirono, i segnali di morte si attenuarono e meno cellule subirono apoptosi.
Cosa significa per i trattamenti futuri
Nel complesso, i risultati delineano una storia coerente: la mutazione GJB2 c.109G>A compromette lo sviluppo e la funzione dell’orecchio interno non solo alterando la comunicazione cellulare, ma attivando anche stress mitocondriale. Questo stress aumenta l’espressione di IFI27 e geni correlati, provoca il rilascio di citocromo c e attiva una cascata di proteine che spinge le cellule ciliate verso la morte programmata. Poiché le cellule ciliate non si rigenerano facilmente negli esseri umani, la loro perdita conduce a deficit uditivi permanenti. Mostrando che la riduzione di IFI27 può attenuare questa cascata distruttiva in cellule umane, lo studio mette in luce IFI27 come un promettente bersaglio per farmaci o terapie geniche. Sebbene tali terapie siano ancora lontane — e probabilmente dovranno essere somministrate molto precocemente nella vita — questo lavoro fornisce una mappa molecolare concreta per trasformare una mutazione genetica un tempo misteriosa in una possibile causa prevenibile di sordità infantile.
Citazione: Chen, Y., Zhao, P., Lin, Q. et al. GJB2 c.109G > A mutation activating IFI27-mediated mitochondrial apoptosis pathway leading to hereditary non-syndromic hearing loss. Sci Rep 16, 6240 (2026). https://doi.org/10.1038/s41598-026-37393-2
Parole chiave: ipoacusia genetica, mutazione GJB2, modello di zebrafish, apoptosi mitocondriale, IFI27