Clear Sky Science · it
Selezione di caratteristiche semplice, veloce ed efficiente guidata da cluster fuzzy adattivi per dati microarray bioinformatici binari ad alta dimensione e fortemente sbilanciati
Perché questo è importante per la ricerca genica
I test moderni di espressione genica possono misurare decine di migliaia di geni in un singolo campione paziente. Questa mole di dati promette diagnosi di cancro più precoci e scelte terapeutiche migliori, ma genera anche un problema: la maggior parte di quei geni è rumorosa, ridondante o legata soprattutto ai casi comuni, non a quelli rari e pericolosi. Questo articolo presenta un nuovo modo per setacciare grandi set di dati di espressione genica in modo che i computer possano identificare in modo affidabile pazienti appartenenti a una piccola minoranza difficile da individuare utilizzando solo un insieme molto ridotto e accuratamente selezionato di geni.
La sfida di troppi geni troppo simili
Gli esperimenti su microarray spesso monitorano migliaia di livelli di attività genica per poche centinaia di pazienti. Di solito, una classe (per esempio un sottotipo di tumore comune) supera di gran lunga l’altra, creando dati fortemente sbilanciati. In questo contesto molti geni si comportano in modo molto simile e i pattern di pazienti della maggioranza e della minoranza possono sovrapporsi. I metodi di apprendimento standard tendono ad aggrapparsi alla classe maggioritaria e a essere confusi dai geni ridondanti, il che porta a overfitting e a una scarsa rilevazione dei sottotipi rari. I metodi tradizionali di riduzione della dimensionalità o perdono interpretabilità costruendo nuove caratteristiche miste, o selezionano geni senza valutare attentamente quanto aiutino un classificatore a riconoscere i casi di minoranza.
Una nuova roadmap per una selezione genica più intelligente
Gli autori introducono AFCG‑SFE, un modello adattivo di selezione delle caratteristiche progettato specificamente per dati di espressione genica ad alta dimensionalità e sbilanciati. Il metodo parte da una semplice ricerca “ad agente singolo” che attiva o disattiva geni e testa quanto supportano la classificazione, ma la arricchisce con diversi passaggi guidati dai dati. Innanzitutto raggruppa i geni in base a quanto si comportano in modo simile, poi permette ai geni di appartenere a più di un gruppo per riflettere la realtà biologica che un gene può essere coinvolto in più vie. All’interno di ciascun gruppo clasifica i geni in base a quanto sono informativi rispetto all’etichetta di malattia e mantiene solo pochi rappresentanti chiave, riducendo drasticamente la ridondanza prima ancora che inizi la ricerca principale. 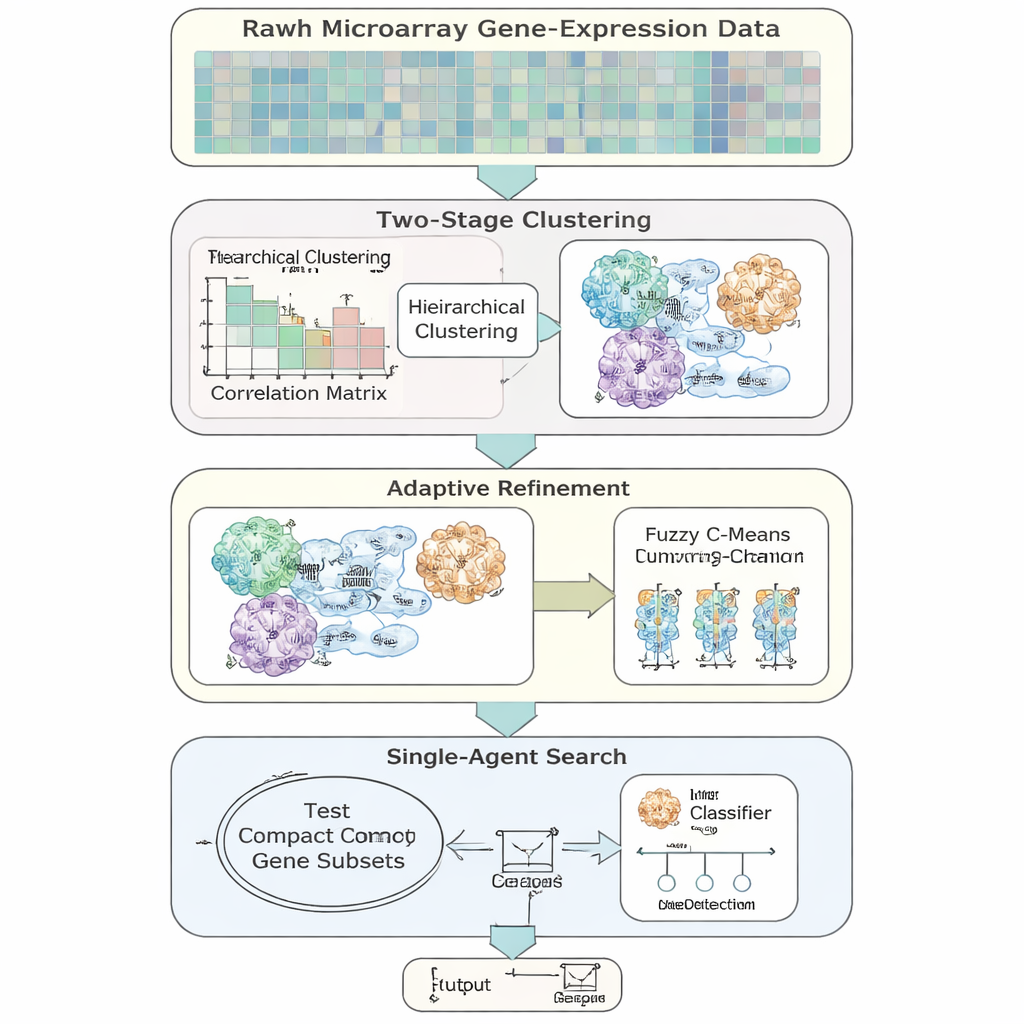
Far sì che il computer dia importanza ai pazienti rari
Invece di concentrarsi sulla semplice accuratezza, AFCG‑SFE usa un punteggio di fitness che enfatizza metriche adatte a dati sbilanciati, inclusi l’equilibrio tra l’identificazione corretta dei casi di minoranza e maggioranza e le prestazioni su tutte le soglie decisionali. La funzione di fitness include anche penalità per la selezione di troppi geni o di molti geni dallo stesso cluster, e un premio per i geni che mostrano una forte dipendenza dall’etichetta di malattia. È importante che l’intensità di queste penalità e ricompense sia impostata automaticamente a partire dalle proprietà del dataset, come il numero di geni per paziente e quanto si sovrappongono le classi, anziché tramite aggiustamenti manuali. Questo rende il metodo più robusto e più facile da trasferire tra studi.
Adattarsi alla difficoltà del problema
Un’idea chiave è che l’algoritmo non debba sempre puntare al set di geni più piccolo possibile. Quando le due classi sono molto difficili da separare o fortemente sovrapposte, il metodo innalza automaticamente un limite inferiore sul numero di geni da mantenere, assicurando che segnali rari ma importanti non vengano scartati. Man mano che la ricerca procede, AFCG‑SFE stringe gradualmente un tetto per cluster sul numero di geni che possono sopravvivere in ogni gruppo, preservando però questo minimo. Il risultato è un pannello compatto e diversificato di geni che cattura la struttura dei dati senza essere dominato da un singolo pattern ridondante. 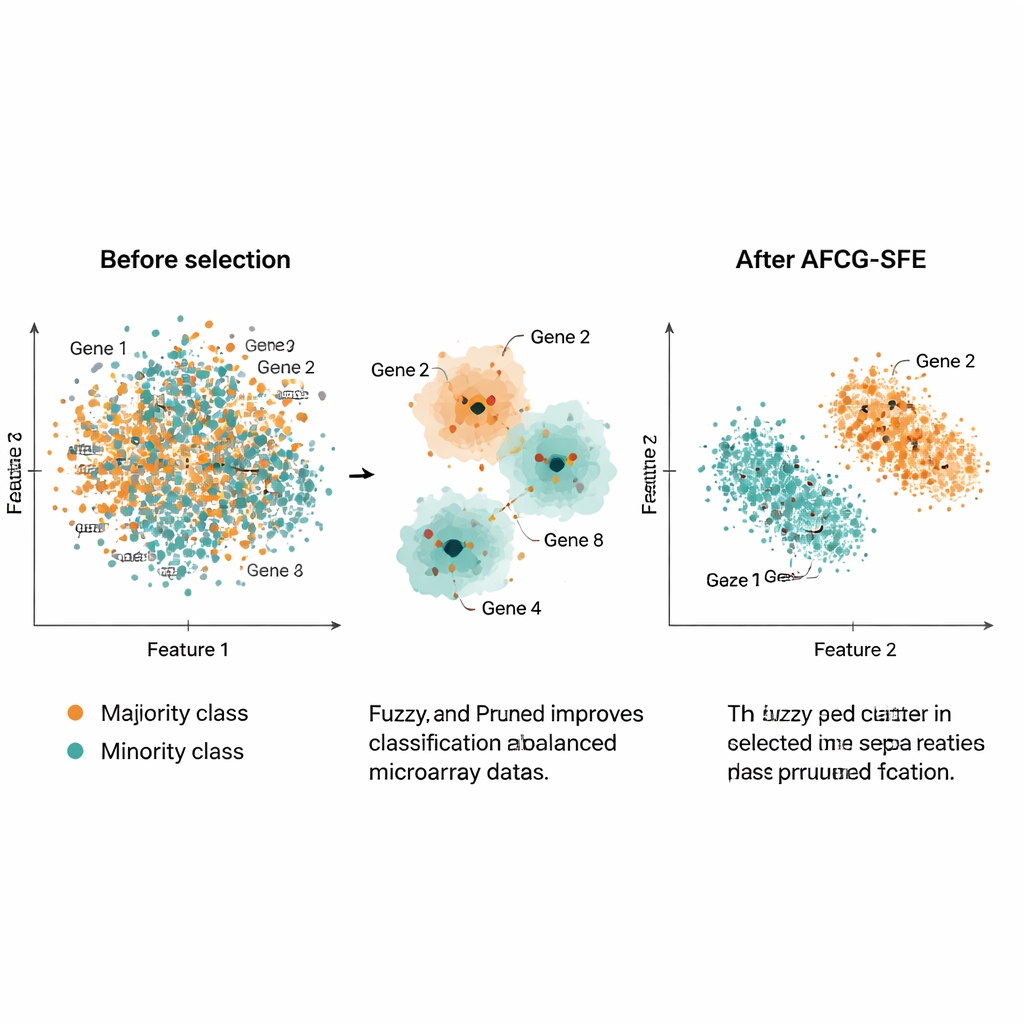
Cosa mostrano gli esperimenti
Gli autori hanno testato AFCG‑SFE su 20 dataset pubblici di microarray tumorali, ciascuno con migliaia di geni ma solo circa 100–200 campioni e un forte sbilanciamento tra le classi. Hanno confrontato il loro metodo con diversi baselines di ricerca evolutiva, filtri semplici e approcci embedded che integrano la selezione delle caratteristiche nel classificatore. Su una batteria di misure — incluse F‑measure, accuratezza bilanciata, area sotto la curva ROC e una misura di overfitting — AFCG‑SFE è risultato il migliore o al pari del migliore su tutti i dataset. Tipicamente ha selezionato meno di 25 geni (spesso solo 6–8), rimuovendo più del 99% delle caratteristiche originali pur migliorando o mantenendo le prestazioni di classificazione. Ha inoltre ridotto un indice di complessità che cattura quanto le classi si sovrappongono nello spazio delle caratteristiche, indicando una separazione più chiara dopo la selezione.
Conclusione per i non esperti
In termini pratici, questo lavoro offre un modo per ridurre profili di espressione genica enormi e rumorosi a insiemi molto piccoli di geni informativi che permettono comunque ai computer di riconoscere con precisione sottogruppi di pazienti rari. Raggruppando in modo intelligente geni simili, premiando quelli che realmente seguono la malattia e proteggendo esplicitamente dalla distorsione verso la classe maggioritaria, AFCG‑SFE fornisce sia previsioni migliori sia pannelli genici molto più semplici. Questa combinazione può aiutare i ricercatori a individuare potenziali biomarcatori, progettare test diagnostici più interpretabili e, in ultima analisi, migliorare il modo in cui gli strumenti di medicina di precisione lavorano con dati biologici reali e imperfetti.
Citazione: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Parole chiave: espressione genica, selezione delle caratteristiche, dati sbilanciati, microarray, sottotipi di cancro