Clear Sky Science · it
rhinotypeR consente l'assegnazione riproducibile del genotipo del rinovirus da sequenze VP4/2
Perché i minuscoli virus del raffreddore contano ancora
La maggior parte di noi considera il comune raffreddore una seccatura piuttosto che una minaccia seria. Eppure i virus che causano molti raffreddori—i rinovirus umani—sono anche collegati a infezioni polmonari gravi, attacchi d'asma e riacutizzazioni di malattie polmonari croniche. Per seguire come questi virus evolvono e si diffondono, gli scienziati devono classificarli in precisi «tipi» genetici, un po' come assegnare codici a barre ai prodotti. Questo articolo presenta rhinotypeR, un pacchetto software gratuito e open source che rende questa etichettatura genetica più accurata, coerente e facilmente ripetibile, aiutando le squadre di sanità pubblica a tenere sotto controllo una famiglia di virus respiratori spesso trascurata.
La varietà nascosta nei comuni raffreddori
I rinovirus umani sono estremamente diffusi, presenti in fino al 60% dei campioni provenienti da persone con malattia respiratoria acuta. Lungi dall'essere un singolo virus, si dividono in tre gruppi principali, chiamati A, B e C, e in almeno 169 tipi genetici riconosciuti. I diversi tipi si comportano in modo diverso: alcuni sono più spesso associati a infezioni gravi nei bambini e a esacerbazioni d'asma, mentre altri compaiono meno frequentemente in malattie severe. Poiché questi tipi evolvono indipendentemente e possiedono caratteristiche superficiali distinte, gli scienziati hanno bisogno di metodi affidabili per distinguerli se vogliono seguire come i focolai si muovono nelle scuole, nelle famiglie e nelle comunità.
Da strumenti sparsi a un unico percorso chiaro
Fino ad ora, l'assegnazione di un tipo di rinovirus dal suo codice genetico è stata un'operazione frammentaria. I ricercatori si concentravano tipicamente su un breve tratto del genoma virale chiamato regione VP4/2, lo allineavano con ceppi di riferimento noti, misuravano quanto differivano le sequenze e poi applicavano valori soglia per decidere a quale tipo apparteneva ogni campione. Ma questi passaggi venivano eseguiti con una miscela di programmi software, editing manuale e giudizio personale. Ciò rendeva difficile confrontare o ripetere diversi studi, anche quando i dati erano simili. rhinotypeR è stato creato proprio per trasformare questo processo a più fasi e incline agli errori in un unico flusso di lavoro scriptato che chiunque può eseguire e condividere.
Cosa fa concretamente il nuovo software
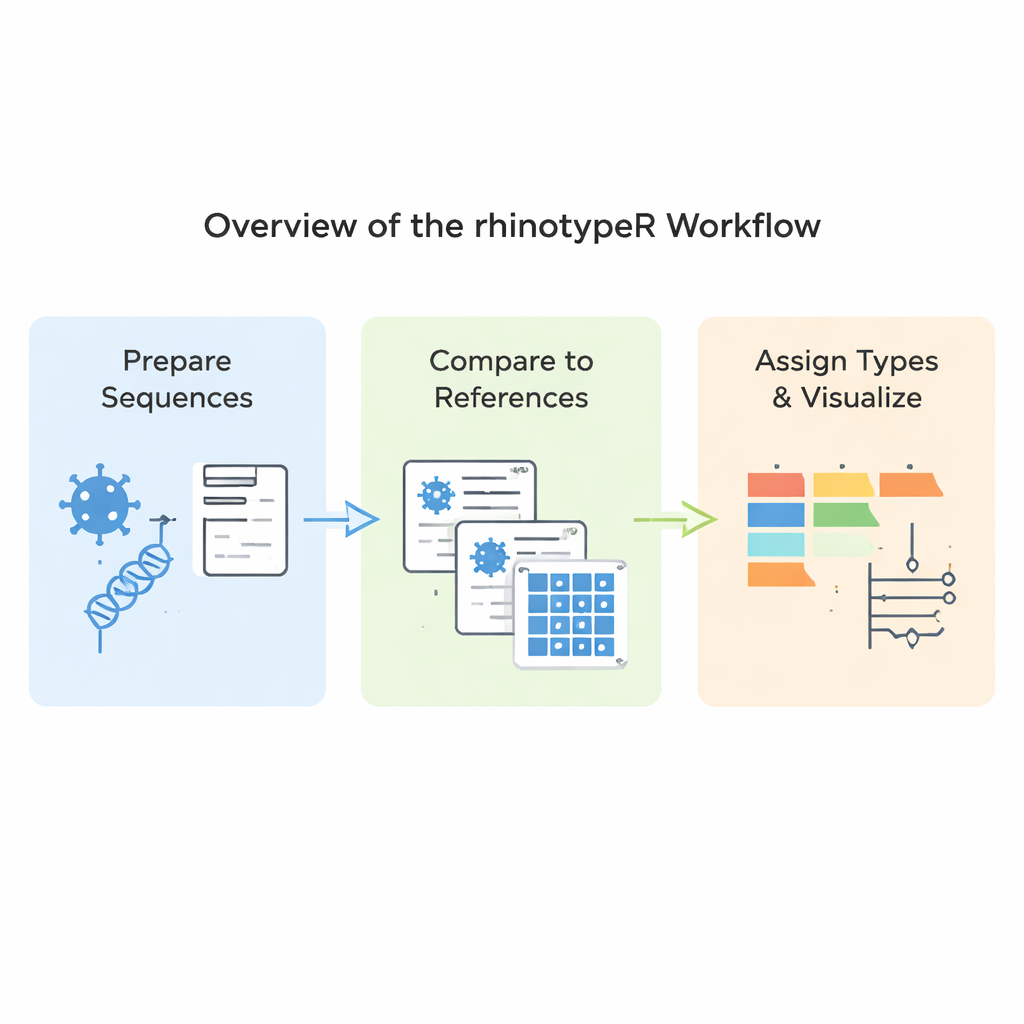
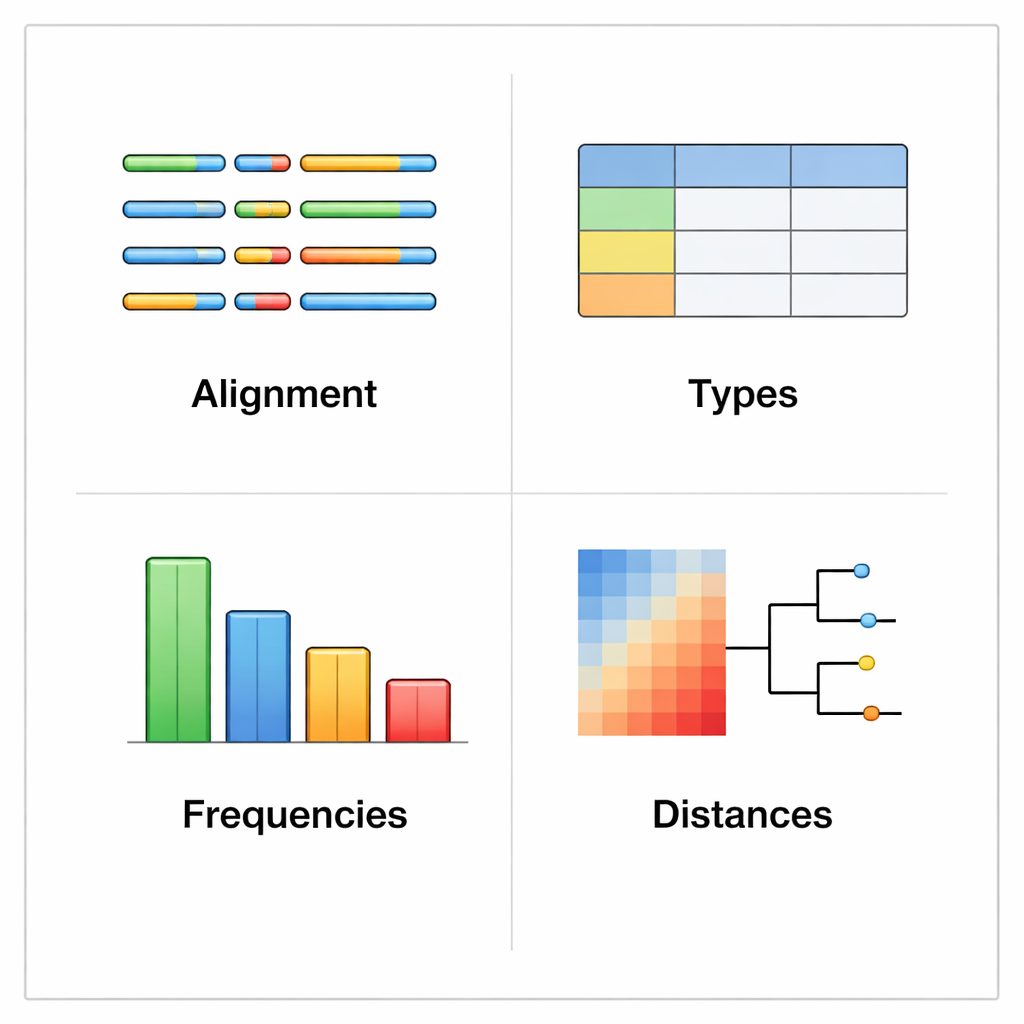
rhinotypeR gira all'interno dell'ambiente R e Bioconductor, ampiamente usato per l'analisi dei dati. Accetta in input una raccolta di sequenze VP4/2 di rinovirus e le porta attraverso tre fasi principali: preparazione e allineamento delle sequenze, calcolo delle distanze di ciascuna rispetto a un set curato di tipi di riferimento e quindi assegnazione di ogni campione al tipo noto più vicino o segnalazione come «non assegnato» se è troppo diverso. Lo stesso strumento può produrre output visivi, inclusi mappe a colori delle differenze genetiche, alberi filogenetici semplici e grafici che mostrano la frequenza di ciascun tipo in un dataset. Gli utenti possono allineare i propri dati con programmi esterni se lo preferiscono, oppure lasciare che rhinotypeR gestisca l'intero processo all'interno di R per la massima riproducibilità.
Mettere lo strumento alla prova
Per verificare che rhinotypeR fornisca risultati affidabili, gli autori hanno confrontato le sue misure di distanza con quelle di due programmi consolidati, ape e MEGA X, usando gli stessi file di input e gli stessi modelli. I risultati sono corrisposti quasi perfettamente; le piccole discrepanze riscontrate erano dovute all'arrotondamento normale nell'aritmetica del computer, non a differenze reali nel metodo. Il team ha poi eseguito rhinotypeR su un'ampia raccolta di oltre 2.300 sequenze di rinovirus provenienti da studi precedenti, coprendo oltre il 90% dei tipi noti. In circa quattro casi su cinque, il nuovo strumento ha concordato esattamente con le etichette di tipo precedenti. La maggior parte dei disaccordi è avvenuta proprio intorno ai punti di soglia predefiniti usati per separare un tipo dall'altro, dove è prevedibile ottenere chiamate borderline. È importante sottolineare che i campioni che non hanno potuto essere assegnati con sicurezza a un tipo noto non mostravano segnali di scarsa qualità o di bassa carica virale, suggerendo che potrebbero riflettere una reale diversità virale.
Perché questo è importante per la sanità pubblica
Per i non specialisti, il messaggio chiave è che rhinotypeR non rivoluziona il modo in cui gli scienziati classificano i virus del raffreddore; piuttosto, rende quel processo più chiaro, trasparente e ripetibile. Raggruppando allineamento, calcoli di distanza e assegnazione dei tipi in un unico pacchetto open source—assieme a riepiloghi visivi chiari—aiuta ricercatori e programmi di sorveglianza a processare migliaia di campioni in modo coerente. Questa coerenza migliora la nostra capacità di confrontare studi provenienti da luoghi e tempi diversi, individuare precocemente linee virali insolite o emergenti e collegare i modelli genetici alle tendenze di malattia nel mondo reale. A lungo termine, strumenti come rhinotypeR rafforzano il monitoraggio di routine di raffreddori apparentemente ordinari che, in molte persone, possono scatenare malattie gravi.
Citazione: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Parole chiave: genotipizzazione del rinovirus, sorveglianza molecolare, sequenziamento VP4/2, strumenti di bioinformatica, virus respiratori