Clear Sky Science · it
Valutazione comparativa dei metodi di profilazione trascrittomica mirata HTG e TempO-Seq
Perché è importante per la cura del cancro
Quando medici e ricercatori studiano il cancro, spesso si rivolgono alle “molecole messaggere” della cellula — l’RNA — per vedere quali geni sono attivi o silenti. Questi schemi possono rivelare come si comporta un tumore e quali terapie potrebbero funzionare meglio. Ma la maggior parte dei campioni ospedalieri viene conservata in blocchi di paraffina dopo fissazione con formalina, che danneggia l’RNA fragile. Questo studio pone una domanda pratica con grandi conseguenze per la ricerca oncologica: ora che un test RNA ampiamente usato è scomparso dal mercato, può un metodo più recente sostituirlo e fornire risultati altrettanto utili a partire da questi campioni conservati di routine?
Due strumenti per leggere l’attività genica
Per anni molti laboratori hanno fatto affidamento su un metodo chiamato HTG EdgeSeq Human Transcriptome Panel (HTP) per leggere l’attività genica direttamente da piccoli raschiati di tessuto fissato in formalina e incluso in paraffina (FFPE). Questo approccio poteva sondare quasi tutti i geni umani senza dover prima estrarre l’RNA, risparmiando tempo e preservando materiale prezioso. Tuttavia, l’azienda dietro HTG EdgeSeq è fallita, lasciando i ricercatori alla ricerca di un’alternativa. Una tecnologia più recente, TempO-Seq (TOS), di un altro produttore, promette capacità simili: anch’essa mira a molti geni in contemporanea, funziona su RNA danneggiato da campioni FFPE ed è progettata per essere sensibile, riproducibile e relativamente economica.
Mettere i metodi alla prova
Il team di ricerca ha confrontato queste due tecnologie testa a testa in un contesto molto pratico. Hanno analizzato 21 campioni conservati di carcinoma endometriale, insieme a tre materiale di riferimento RNA standard, prima con HTG HTP e poi con TempO-Seq. Entrambi i metodi hanno utilizzato pannelli mirati che complessivamente coprivano più di 18.000 degli stessi geni. Gli scienziati hanno applicato controlli di qualità rigorosi, assicurandosi che ogni campione producesse un numero sufficiente di letture di sequenziamento e che le misure fossero stabili. Hanno inoltre usato strumenti statistici per rimuovere gli “effetti batch” — differenze artificiali che possono emergere semplicemente perché i test sono stati eseguiti in giorni, macchine o piattaforme diverse.
Cosa corrisponde e cosa no
Quando il gruppo ha esaminato l’espressione dei singoli geni uno per uno, i due metodi non concordavano sempre. Differenze nel modo in cui ciascuna tecnologia progetta le sonde, prepara i campioni e conta le letture possono rendere le comparazioni gene-singolo rumorose. Tuttavia, questo quadro è cambiato quando hanno esaminato schemi più ampi che combinano informazioni provenienti da molti geni contemporaneamente. Le firme multi-geniche — come quelle usate per raggruppare i tumori in sottotipi molecolari, stimare quante cellule immunitarie sono presenti in un campione o valutare la purezza del tessuto tumorale — hanno mostrato un accordo molto più forte tra TempO-Seq e HTG. Nella maggior parte dei casi, i punteggi o le classificazioni erano simili, anche dopo che i ricercatori avevano simulato l’uso di un numero inferiore di letture di sequenziamento per imitare diverse capacità delle macchine.
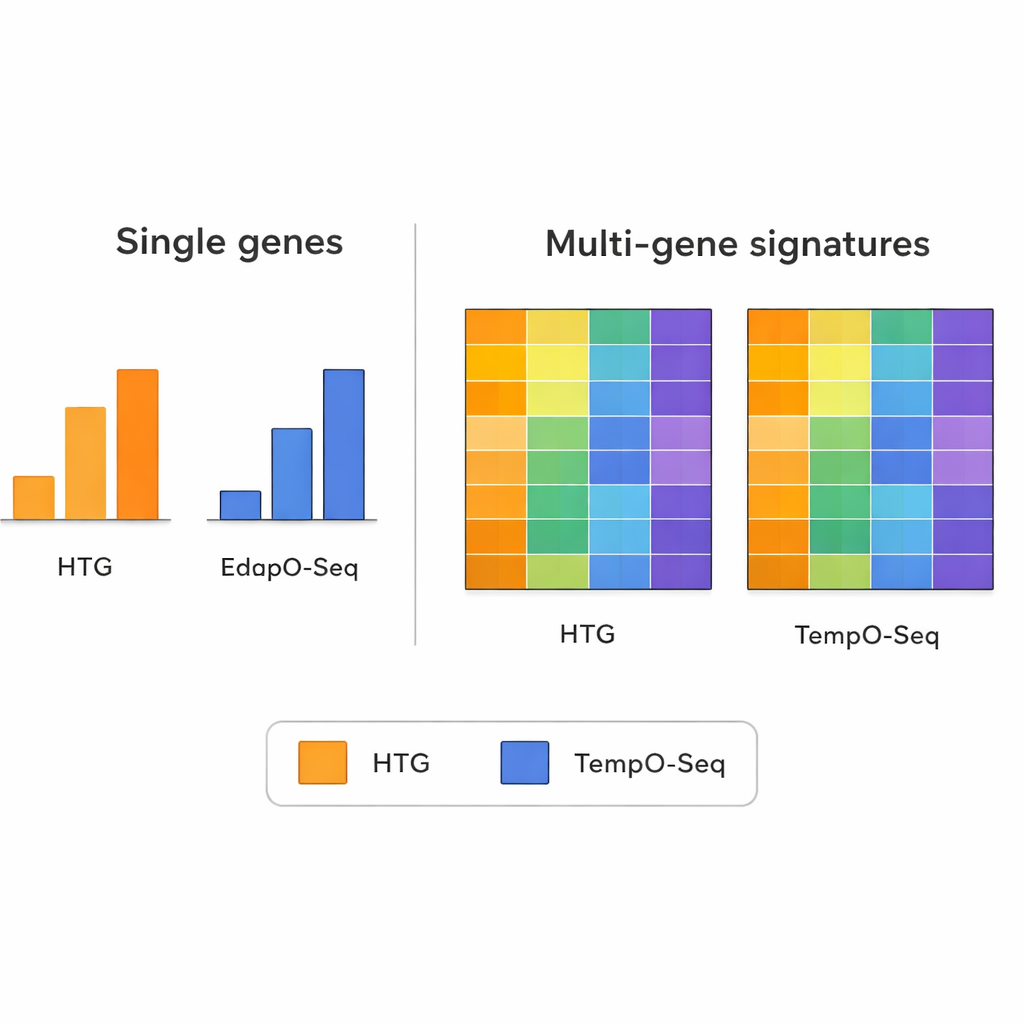
Le firme multi-geniche come segnali affidabili
Lo studio evidenzia un principio importante nella genomica moderna: mentre la misura di un singolo gene può essere falsata da inghippi tecnici, combinare segnali da dozzine o centinaia di geni tende a compensare quel rumore. Gli autori hanno utilizzato diversi strumenti multi-genici noti come test di stress tecnico. Questi includevano un pannello per il carcinoma mammario che assegna i tumori a sottotipi intrinseci, un algoritmo che valuta quanto tessuto immunitario e connettivo sia mescolato in un campione tumorale e un metodo che stima le proporzioni di molti tipi di cellule immunitarie. In questi complessi profili, TempO-Seq in genere ha seguito da vicino HTG, suggerendo che cattura le stesse storie biologiche anche se alcuni dettagli fini differiscono.
Cosa significa per il futuro
Per i ricercatori che dipendono dagli archivi FFPE per studiare il cancro, la perdita di una piattaforma affidabile avrebbe potuto rappresentare un grosso ostacolo. Questo studio di benchmarking offre rassicurazioni: TempO-Seq sembra essere una sostituzione solida per HTG HTP quando l’obiettivo è usare biomarcatori multi-genici e modelli di espressione ampi, che sono la spina dorsale di molti strumenti diagnostici e prognostici moderni. Gli autori avvertono che confrontare direttamente i risultati di un singolo gene tra piattaforme è sconsigliabile, perché ogni metodo mira ai geni in modi leggermente diversi. Raccomandano invece di concentrarsi su firme complesse multi-geniche per lavori cross-piattaforma. In termini semplici, il nuovo metodo sembra in grado di proseguire il lavoro del suo predecessore per la maggior parte delle necessità di ricerca oncologica nel mondo reale, specialmente quando gli scienziati si interessano al quadro complessivo di molti geni piuttosto che al valore esatto di uno solo.
Citazione: Fernández-Serra, A., López-Reig, R., Romero, I. et al. Comparative evaluation of HTG and TempO Seq targeted transcriptome profiling methods. Sci Rep 16, 6108 (2026). https://doi.org/10.1038/s41598-026-36810-w
Parole chiave: profilazione trascrittomica, carcinoma endometriale, tessuto FFPE, sequenziamento RNA mirato, biomarcatori di espressione genica