Clear Sky Science · it
Un approccio di machine learning per prevedere i coefficienti osmotici e ricavare i coefficienti di attività nei sali di ammonio alchilici
Sostanze di tutti i giorni con complessità nascosta
Dai ammorbidenti per tessuti e i balsami per capelli alle salviette disinfettanti e al collutorio, una famiglia di sostanze chiamate sali di ammonio quaternario—spesso abbreviati in “Quat”—è silenziosamente alla base di molti prodotti su cui facciamo affidamento. Aiutano a uccidere i germi, ammorbidire i tessuti e accelerare reazioni industriali. Tuttavia prevedere esattamente come questi sali si comportano in acqua si è rivelato sorprendentemente difficile, limitando l’efficienza con cui possiamo progettare formulazioni più sicure e più ecologiche. Questo studio mostra come il machine learning moderno può apprendere dai dati precedenti per prevedere tale comportamento in modo più flessibile e, in molti casi, più accurato rispetto ai modelli tradizionali.
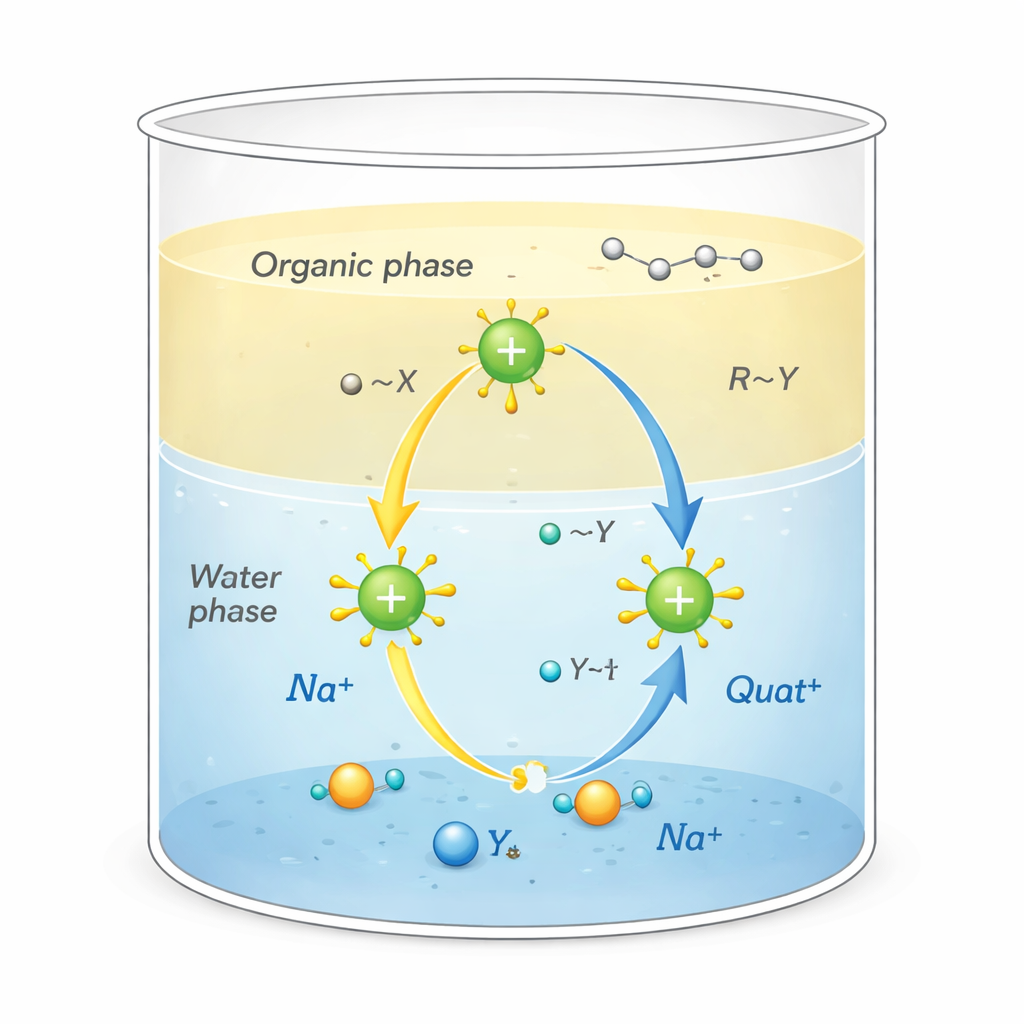
Perché questi sali sono importanti
I Quat sono molecole cariche positivamente circondate da “code” ricche di carbonio. Questa forma insolita permette loro di svolgere più funzioni contemporaneamente: aderire allo sporco oleoso, attaccarsi a superfici come tessuti o capelli e perturbare le membrane dei microrganismi, rendendoli potenti disinfettanti e tensioattivi. Sono usati anche come catalizzatori di trasferimento di fase, agendo come navette che trasportano ioni reattivi dall’acqua verso solventi oleosi dove normalmente non si disperderebbero. Questa azione di trasporto, che avviene alla frontiera tra acqua e olio, può accelerare drasticamente reazioni chimiche impiegate nella produzione di farmaci, polimeri e prodotti chimici di pregio.
Perché è difficile prevederne il comportamento
Per progettare nuovi Quat o ottimizzarne di esistenti, i chimici devono conoscere come si comportano in soluzione—quanto interagiscono con l’acqua e con altri ioni disciolti. Due misure chiave sono il coefficiente osmotico, che riflette come i sali influenzano la tendenza dell’acqua a migrare attraverso membrane, e il coefficiente di attività, che cattura quanto una specie disciolta è “efficace” rispetto a una soluzione ideale perfettamente mescolata. Tradizionalmente, questi valori si ottengono tramite esperimenti meticolosi o usando modelli fisici complessi come Electrolyte‑NRTL e Extended UNIQUAC, che richiedono molti parametri adattati e non si generalizzano facilmente a nuove molecole.
Insegnare a un computer a leggere le molecole
I ricercatori hanno seguito una strada diversa: hanno chiesto se un computer potesse apprendere il legame tra struttura dei Quat e comportamento osmotico direttamente dai dati esistenti. Hanno raccolto 1.654 misure di coefficienti osmotici per 52 diversi Quat dalla letteratura scientifica. Ogni molecola è stata descritta usando la notazione SMILES—una rappresentazione testuale che codifica caratteristiche come il numero di atomi di carbonio e ossigeno, la presenza di anelli benzenici, il grado di ramificazione e il tipo di gruppo azotato caricato positivamente, insieme all’anione accompagnatore (come cloruro, bromuro o nitrato). Questi descrittori strutturali, insieme alla concentrazione del sale, sono stati usati come input per diversi algoritmi di apprendimento supervisionato implementati in Python.
Trovare il predittore più affidabile
Sette algoritmi differenti, tra cui regressione lineare, alberi decisionali, foreste casuali, macchine a vettori di supporto, gradient boosting, k‑nearest neighbors e processi gaussiani, sono stati addestrati sul 70% dei dati e testati sul restante 30%. Il team ha anche adottato uno schema di validazione più rigoroso in cui tutti i dati relativi a un singolo sale erano esclusi per valutare quanto bene i modelli si potessero estendere a un composto veramente non visto. La regressione lineare ha reso male, non cogliendo importanti tendenze non lineari. I metodi basati su alberi hanno adattato molto bene i dati di addestramento ma hanno prodotto previsioni leggermente frastagliate e hanno perso accuratezza su sali nuovi. Il modello a processo gaussiano ha trovato il miglior compromesso: ha fornito curve lisce e fisicamente ragionevoli per i coefficienti osmotici e ha raggiunto un errore percentuale assoluto medio di circa il 5% complessivo, superando gli altri approcci di machine learning nei test più severi.
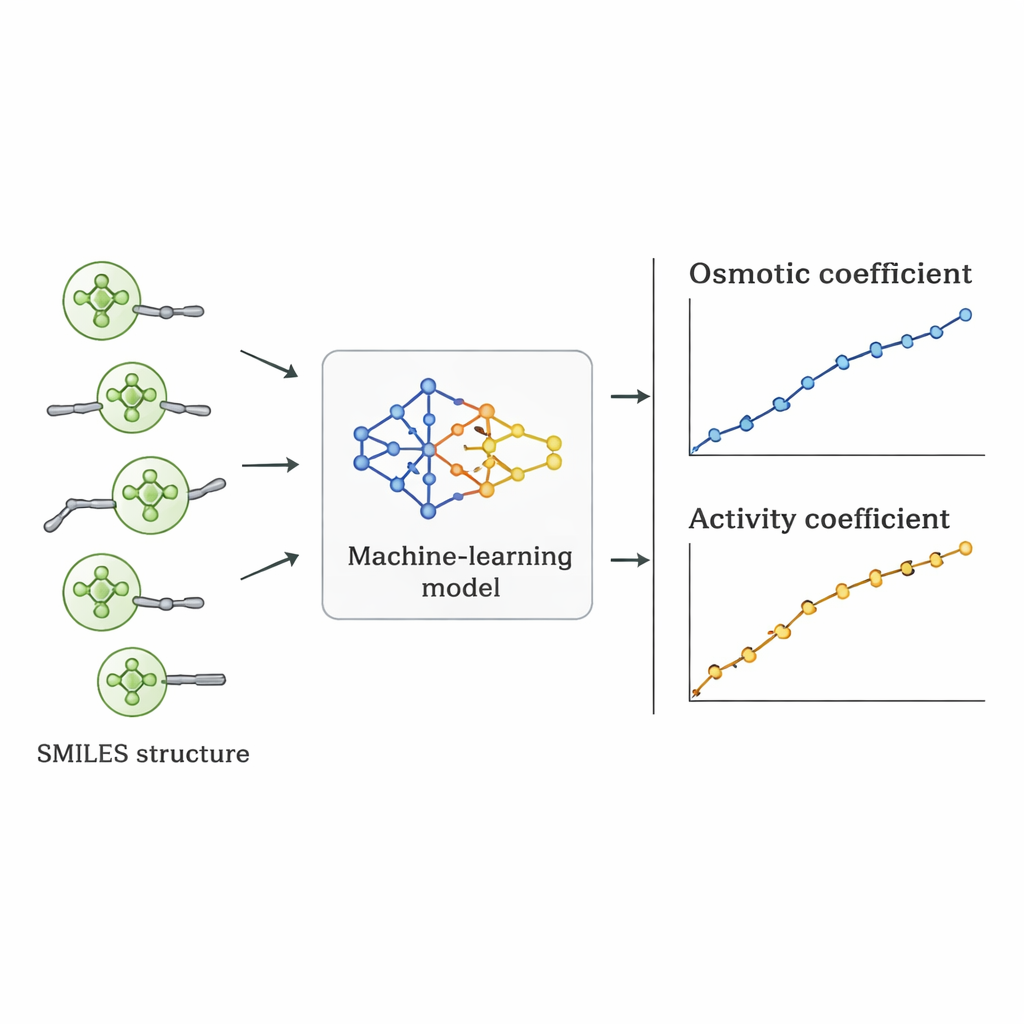
Dal comportamento osmotico a numeri utili per il progetto
Una volta scelto il miglior modello, i coefficienti osmotici predetti sono stati convertiti in coefficienti di attività usando relazioni termodinamiche standard. Quando questi coefficienti di attività sono stati confrontati con valori derivati da esperimenti e da modelli fisici consolidati, l’approccio basato sui dati spesso li ha eguagliati o superati per singoli Quat. Sebbene l’errore medio su tutte le sostanze fosse leggermente superiore rispetto ad alcuni modelli specializzati, presentava un vantaggio cruciale: poiché è guidato da descrittori strutturali piuttosto che da adattamenti specifici per ogni sale, può essere applicato a nuovi Quat mai misurati in laboratorio, a condizione che le loro strutture siano simili a quelle del set di addestramento.
Cosa significa per prodotti e processi
Per un non specialista, il messaggio è che i computer possono ora “leggere” descrizioni testuali compatte di molecole e, dai modelli appresi sui dati del passato, prevedere con notevole accuratezza come queste molecole si comporteranno in acqua. Questo apre la strada a uno screening più rapido e meno costoso di nuovi Quat per disinfettanti, detergenti, prodotti per la cura personale e catalizzatori industriali, senza esperimenti esaustivi per ogni candidato. Il modello attuale è solo un primo passo, e gli autori osservano che impronte molecolari più ricche e algoritmi più recenti potrebbero migliorare ulteriormente le prestazioni. Tuttavia dimostra come gli strumenti guidati dai dati possano integrare la chimica tradizionale, aiutando gli ingegneri a progettare formulazioni più efficaci e potenzialmente più sicure esplorando possibilità chimiche che sarebbe poco pratico testare una per una in laboratorio.
Citazione: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Parole chiave: sali di ammonio quaternario, catalisi di trasferimento di fase, coefficienti osmotici, coefficienti di attività, machine learning in chimica