Clear Sky Science · it
Quadro di apprendimento automatico per l’analisi dello splicing alternativo dell’mRNA identifica una firma di progressione nell’adenocarcinoma colorettale
Perché questa ricerca conta per i pazienti
Il cancro colorettale è uno dei tumori più comuni e letali, e tuttavia i medici faticano ancora a prevedere quali tumori resteranno silenziosi e quali torneranno aggressivi dopo il trattamento. Questo studio presenta un nuovo modo di leggere segnali nascosti nell’RNA tumorale — i messaggi che le cellule usano per produrre proteine — e utilizza l’apprendimento automatico per trasformare questi segnali in un semplice punteggio di rischio che potrebbe aiutare a modulare quanto aggressivamente trattare ciascun paziente.
Tagli ed edit nascosti nei geni del cancro
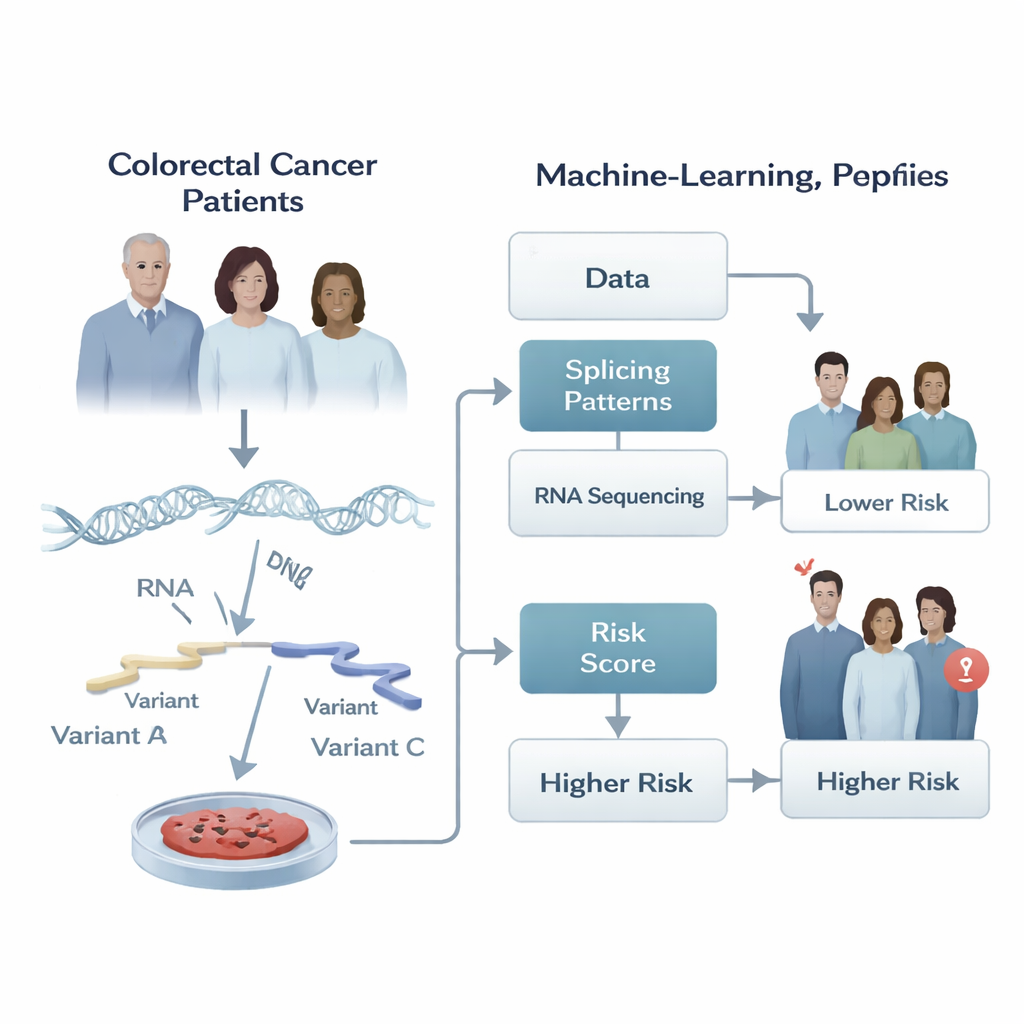
I nostri geni non vengono letti in modo fisso. Quando una cellula copia il DNA in RNA, può tagliare e incollare segmenti del messaggio RNA in combinazioni diverse, un processo chiamato splicing alternativo. Questo editing permette a un singolo gene di produrre più versioni di una proteina, come diversi attrezzi nello stesso kit. Nelle cellule sane questa flessibilità è strettamente regolata. Nel cancro, però, il taglio e l’incollaggio possono andare storti, generando versioni di proteine che favoriscono la crescita del tumore, la sua diffusione e la resistenza alle terapie. Gli autori hanno ipotizzato che il pattern di questi edit dell’RNA in un tumore possa contenere indizi importanti sul comportamento che quel cancro avrà nel tempo.
Trasformare i pattern di RNA in un punteggio di rischio
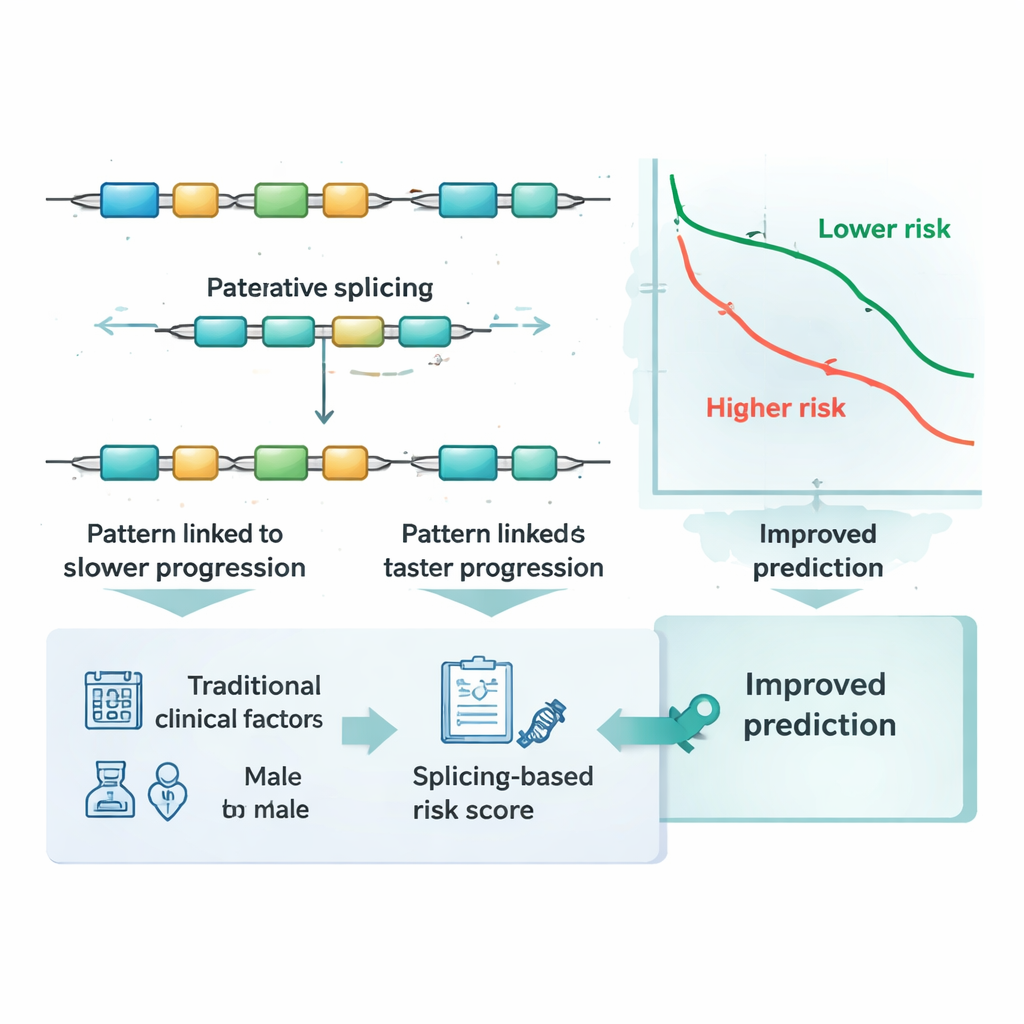
I ricercatori hanno analizzato dati di sequenziamento RNA provenienti dai tumori di 266 pazienti con adenocarcinoma colorettale nel The Cancer Genome Atlas e di altri 348 pazienti in uno studio indipendente. Per ogni tumore hanno quantificato quanto spesso venivano utilizzate scelte di splicing particolari, riassumendole con un numero compreso tra zero e uno. Hanno quindi costruito una pipeline di apprendimento automatico a più fasi che prima ha filtrato migliaia di eventi di splicing per individuare eventuali legami con il tempo libero da progressione del tumore, e poi ha ridotto attentamente questa lista evitando segnali ridondanti o sovrapposti. Il risultato finale è stato una ‘‘firma’’ compatta di appena cinque eventi di splicing specifici il cui comportamento combinato seguiva al meglio se il cancro di un paziente progrediva prima o dopo.
Dividere i pazienti in gruppi a rischio più basso e più alto
Usando questa firma di cinque eventi, il team ha definito un punteggio di rischio numerico per ogni paziente sommando le misurazioni di splicing, ponderate in base a quanto ciascuna fosse correlata alla progressione. I pazienti i cui tumori presentavano tre dei pattern di splicing avevano tendenzialmente esiti peggiori, mentre due pattern erano associati a risultati migliori. Il punteggio ha diviso nettamente i pazienti in gruppi a basso e alto rischio: sia nella coorte originale sia nel gruppo di validazione indipendente, chi aveva punteggi elevati ha mostrato una progressione del cancro significativamente più precoce. Quando i ricercatori hanno tracciato le curve del tempo alla progressione, le due linee si sono separate chiaramente, indicando che questo piccolo insieme di edit dell’RNA coglieva differenze significative nel comportamento tumorale in centinaia di individui.
Oltre la stadiazione standard e i marker noti
I medici attualmente si basano su stadiazione tumorale, età e altre caratteristiche cliniche per stimare il rischio, e talvolta su specifiche alterazioni del DNA o sui livelli di espressione genica. I ricercatori si sono chiesti se il loro punteggio basato sullo splicing apportasse valore aggiunto rispetto a queste misure consolidate. Utilizzando test di accuratezza dipendenti dal tempo, hanno mostrato che le previsioni basate solo su stadiazione, età e sesso miglioravano in modo evidente quando veniva incluso il punteggio di rischio dello splicing. Hanno inoltre confrontato il punteggio con decine di noti marker molecolari nel cancro colorettale e con diversi approcci statistici comuni. In entrambi i gruppi principali di pazienti, la firma di cinque eventi di splicing ha eguagliato o superato queste alternative e ha migliorato la predizione quando usata insieme a esse, suggerendo che cattura informazioni che altri marker non rilevano.
Cosa potrebbe significare per le cure future
Per un pubblico non specialistico, il messaggio chiave è che il modo in cui un tumore ‘‘edita’’ il suo RNA può rivelare quanto sia probabile che sia pericoloso. Questo studio mostra che monitorare appena cinque edit specifici dell’RNA nei tumori colorettali può suddividere i pazienti in gruppi con differenze significative nella probabilità di rimanere liberi da progressione. Sebbene questo lavoro debba ancora essere tradotto in test di laboratorio pratici e valutato in trial clinici prospettici, indica una direzione in cui i medici potrebbero usare tale punteggio alla diagnosi per decidere chi necessita di terapie più aggressive e sorveglianza più ravvicinata, e chi potrebbe evitare un sovratrattamento. Più in generale, offre un quadro riutilizzabile per analizzare i pattern di splicing dell’RNA in altri tumori al fine di affinare la prognosi e guidare terapie veramente personalizzate.
Citazione: Maimekov, U., Nosrati, M., Mahmoud, A. et al. Machine learning framework for mRNA alternative splicing analysis identifies a signature of progression in colorectal adenocarcinoma. Sci Rep 16, 7106 (2026). https://doi.org/10.1038/s41598-026-35903-w
Parole chiave: cancro colorettale, splicing alternativo, sequenziamento RNA, apprendimento automatico, prognosi del cancro