Clear Sky Science · it
Uno studio quantitativo di composti citotossici usando descrittori basati su grafi e apprendimento automatico
Perché questa ricerca è importante per i farmaci oncologici del futuro
I farmaci antitumorali che uccidono le cellule tumorali, noti come farmaci citotossici, spesso camminano su una lama di raso tra salvare vite e causare effetti collaterali gravi. Per progettare terapie più sicure ed efficaci, gli scienziati hanno bisogno di metodi rapidi e affidabili per prevedere come questi farmaci si muovono nell’organismo—quanto vengono assorbiti, quanto facilmente attraversano le membrane cellulari e dove finiscono. Questo studio mostra come descrizioni matematiche delle molecole, combinate con tecniche moderne di apprendimento automatico, possano stimare con precisione una proprietà chiave che controlla questo comportamento, accelerando potenzialmente la ricerca di terapie oncologiche migliori.
Una superficie chiave che controlla dove i farmaci possono andare
Un’idea centrale nell’articolo è l’area superficiale polare topologica, o Top_PSA. In termini semplici, si tratta di un numero che riflette quanto della superficie di una molecola è costituito da regioni “polari”—parti che amano l’acqua e possono formare legami a idrogeno. Molecole con aree superficiali polari molto elevate spesso faticano ad attraversare le membrane lipidiche e possono essere scarsamente assorbite per via orale. Molecole con aree superficiali polari molto basse possono invece attraversare facilmente molte barriere, a volte causando effetti indesiderati in tessuti sensibili come il cervello. La Top_PSA è diventata un pratico indicatore per stimare queste proprietà di trasporto perché può essere calcolata rapidamente da una rappresentazione 2D della molecola, senza bisogno di lente simulazioni 3D.
Trasformare i disegni molecolari in numeri
I ricercatori hanno assemblato un insieme curato di 156 diversi composti citotossici ricavati da farmaci antitumorali reali e da agenti sperimentali. Hanno quindi convertito ogni molecola in 58 cosiddetti descrittori—numeri che catturano caratteristiche come il numero di atomi, il numero di anelli, la flessibilità dei legami, il numero di atomi in grado di formare legami a idrogeno e quanto sono polari o elettronegativi i diversi gruppi. Molti di questi descrittori derivano dalla teoria dei grafi, in cui una molecola è trattata come una rete di nodi e collegamenti. Questo ricco ritratto numerico di ciascuna molecola è servito come input per modelli al computer che cercano di prevedere i valori di Top_PSA calcolati da popolari toolkit chimici.
Mettere alla prova più strade per una previsione accurata
Per trovare il modo migliore di collegare questi descrittori alla Top_PSA, il gruppo ha confrontato diverse strategie di modellizzazione. Hanno provato la regressione lineare standard e due versioni “regolarizzate” chiamate regressione ridge e LASSO, progettate per gestire meglio informazioni rumorose e sovrapposte. Hanno inoltre esplorato diversi schemi di preparazione dei dati: adattare i modelli direttamente sui descrittori grezzi, comprimerli con l’analisi delle componenti principali (PCA), scalarli in modo da ridurre l’impatto dei valori estremi (robust scaling), gestire gli outlier e potare le feature altamente correlate usando una misura chiamata variance inflation factor. Ogni approccio è stato valutato con cura usando la validazione incrociata k‑fold, un metodo che suddivide ripetutamente i dati in sottoinsiemi di addestramento e test per prevenire l’overfitting.
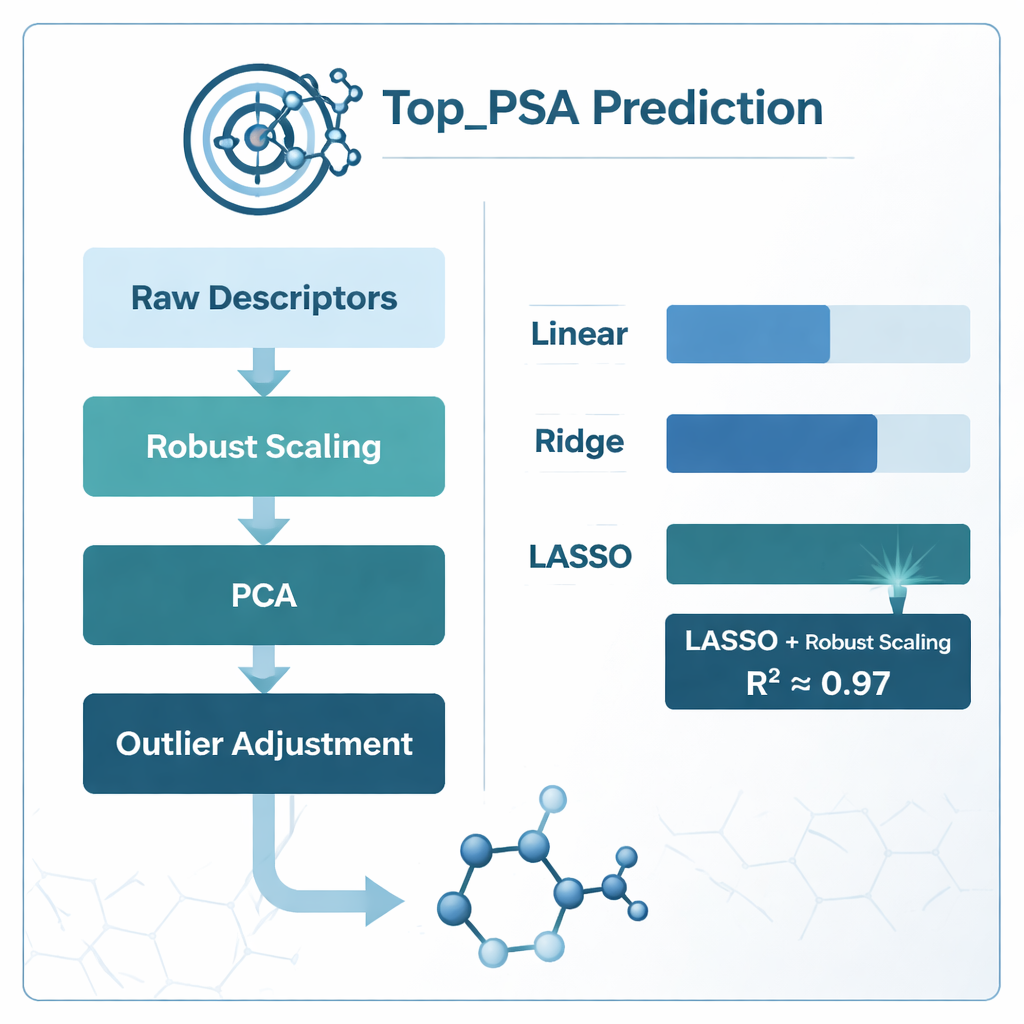
Cosa ha funzionato meglio e cosa hanno imparato i modelli
Il vincitore chiaro è stata la combinazione di robust scaling con la regressione LASSO, che ha raggiunto un coefficiente di determinazione (R²) di circa 0,97—ossia è riuscita a spiegare approssimativamente il 97% della variazione della Top_PSA tra i 156 farmaci. I modelli basati su PCA si sono avvicinati per accuratezza grezza ma erano più difficili da interpretare chimicamente perché i descrittori originali vengono miscelati in componenti astratte. La semplice potatura dei descrittori correlati usando il variance inflation factor ha in realtà peggiorato le prestazioni, suggerendo che alcune misure sovrapposte contengono comunque informazioni chimiche utili. Esaminando quali pesi dei descrittori la LASSO ha mantenuto diversi da zero, gli autori hanno trovato che i fattori più importanti erano la presenza di eteroatomi come azoto e ossigeno, la capacità di donare o accettare legami a idrogeno e indici che tracciano come gli atomi più elettronegativi sono distribuiti nel grafo molecolare—tutte caratteristiche che coincidono con l’intuizione chimica della superficie polare.
Come questo può guidare un migliore design dei farmaci
Per i lettori non specialisti, il messaggio chiave è che impronte matematiche delle molecole preparate con cura, abbinate a metodi di apprendimento automatico ben scelti, possono fornire stime rapide e affidabili di quanto i farmaci oncologici saranno “appiccicosi” o “scivolosi” mentre si muovono nell’organismo. Lo studio offre indicazioni pratiche per altri ricercatori su come preprocessare i dati dei descrittori, quali approcci di modellizzazione privilegiare e quali scorciatoie evitare. A lungo termine, modelli interpretabili e robusti della Top_PSA possono aiutare i chimici a filtrare vaste librerie virtuali di potenziali farmaci, concentrando gli sforzi su composti con il giusto equilibrio tra capacità di attraversare le membrane e sicurezza—un passo importante verso trattamenti anticancro più efficaci e meno tossici.
Citazione: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Parole chiave: farmaci citotossici, area superficiale polare, descrittori molecolari, apprendimento automatico, permeabilità dei farmaci