Clear Sky Science · it
Disfunzione mitocondriale e disregolazione del Ca2+ in neuroni umani derivati da iPSC con mutazione di presenilina‑1 emergono sotto stress tramite un meccanismo indipendente da MCU‑1
Perché questo è importante per la malattia di Alzheimer
La malattia di Alzheimer viene spesso descritta attraverso le placche proteiche appiccicose nel cervello, ma molto prima che la memoria cominci a declinare, le piccole “centrali energetiche” all’interno dei neuroni—i mitocondri—e la gestione degli ioni calcio possono già non funzionare correttamente. Questo studio utilizza neuroni umani coltivati da cellule della pelle di una persona portatrice di una nota mutazione familiare dell’Alzheimer per porre una domanda semplice ma cruciale: quanto presto, e in che modo, la produzione di energia e l’equilibrio del calcio cominciano a guastarsi?
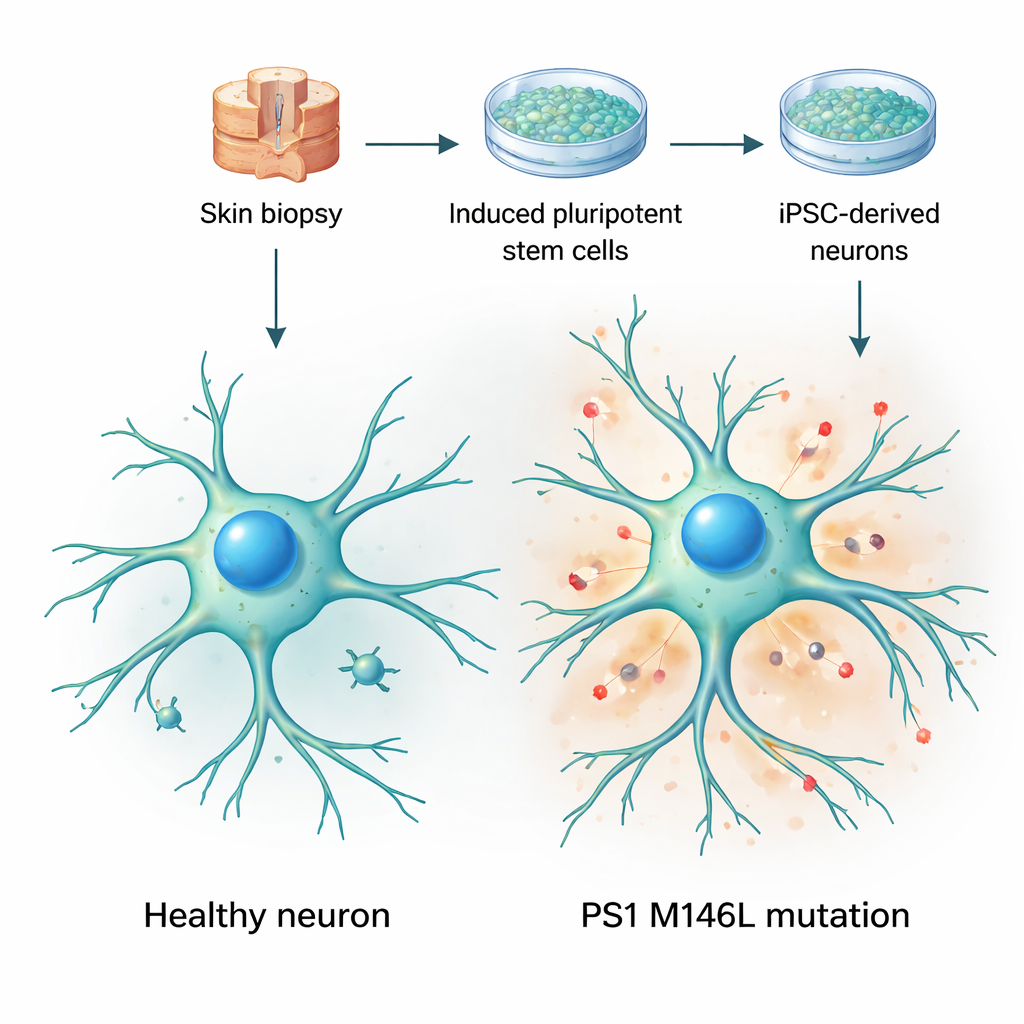
Trasformare le cellule della pelle in modelli cerebrali vivi
I ricercatori sono partiti da biopsie cutanee di due donne: una volontaria sana e una portatrice asintomatica della mutazione di presenilina‑1 chiamata M146L, presente in una famiglia argentina con Alzheimer a insorgenza precoce. Hanno riprogrammato le cellule della pelle in cellule staminali pluripotenti indotte—cellule capaci di differenziarsi in quasi qualsiasi tessuto—e poi le hanno indotte a diventare neuroni. Nel corso di diverse settimane in coltura, queste cellule hanno acquisito morfologie neuronali tipiche, esteso lunghi processi ramificati ed espresso marcatori neuronali standard. Importante, sia le cellule di controllo sia quelle mutanti hanno maturato a ritmi simili e presentavano un aspetto globalmente sano, permettendo al gruppo di concentrarsi su cambiamenti funzionali sottili anziché su perdita o danno cellulare evidente.
Segnali elettrici e calcio sotto stress
I neuroni dipendono da un controllo rigoroso del calcio, un atomo carico che funge da interruttore rapido per molti processi cellulari. Utilizzando coloranti fluorescenti, il team ha seguito come cambiavano i livelli di calcio all’interno delle cellule quando queste venivano stimolate elettricamente con potassio o attivate con molecole segnalatrici. Sotto una semplice stimolazione depolarizzante, i neuroni portatori della mutazione M146L mostravano incrementi di calcio più deboli rispetto ai neuroni di controllo, suggerendo difficoltà nel mantenere i gradienti elettrici e ionici che normalmente guidano l’ingresso del calcio. Tuttavia, quando i ricercatori hanno innescato una situazione più stressante—costringendo il calcio a fuoriuscire dai depositi interni del reticolo endoplasmatico—la differenza è diventata più evidente. In risposta a questo stress, i mitocondri dei neuroni mutanti hanno assorbito una quantità di calcio nettamente inferiore rispetto a quelli di controllo, indicando una ridotta capacità di smorzare pericolosi picchi di calcio.
Disaccoppiamento tra uso di energia ed equilibrio del calcio
Per capire come questa alterata gestione del calcio influenzi il metabolismo cellulare, gli investigatori hanno misurato quanto ossigeno consumavano i neuroni—un proxy diretto dell’attività mitocondriale. Sorprendentemente, i neuroni con la mutazione M146L consumavano più ossigeno: i loro tassi di consumo basale e massimale di ossigeno, e la quantità di ossigeno collegata alla produzione di ATP, erano tutti più elevati rispetto alle cellule di controllo. Eppure l’efficienza del legame tra consumo di ossigeno e produzione di ATP risultava simile, e non c’era un aumento del numero di mitocondri né degli enzimi chiave per la sintesi di ATP. Invece, i mitocondri nei neuroni mutanti erano più lunghi e tubulari, con livelli più alti di una proteina di fusione chiamata mitofusina‑1, uno schema spesso osservato in cellule sottoposte a stress cronico di basso grado. Questi mitocondri iperattivi ed allungati generavano anche più specie reattive dell’ossigeno, molecole instabili che possono danneggiare proteine e DNA se non vengono adeguatamente controllate.
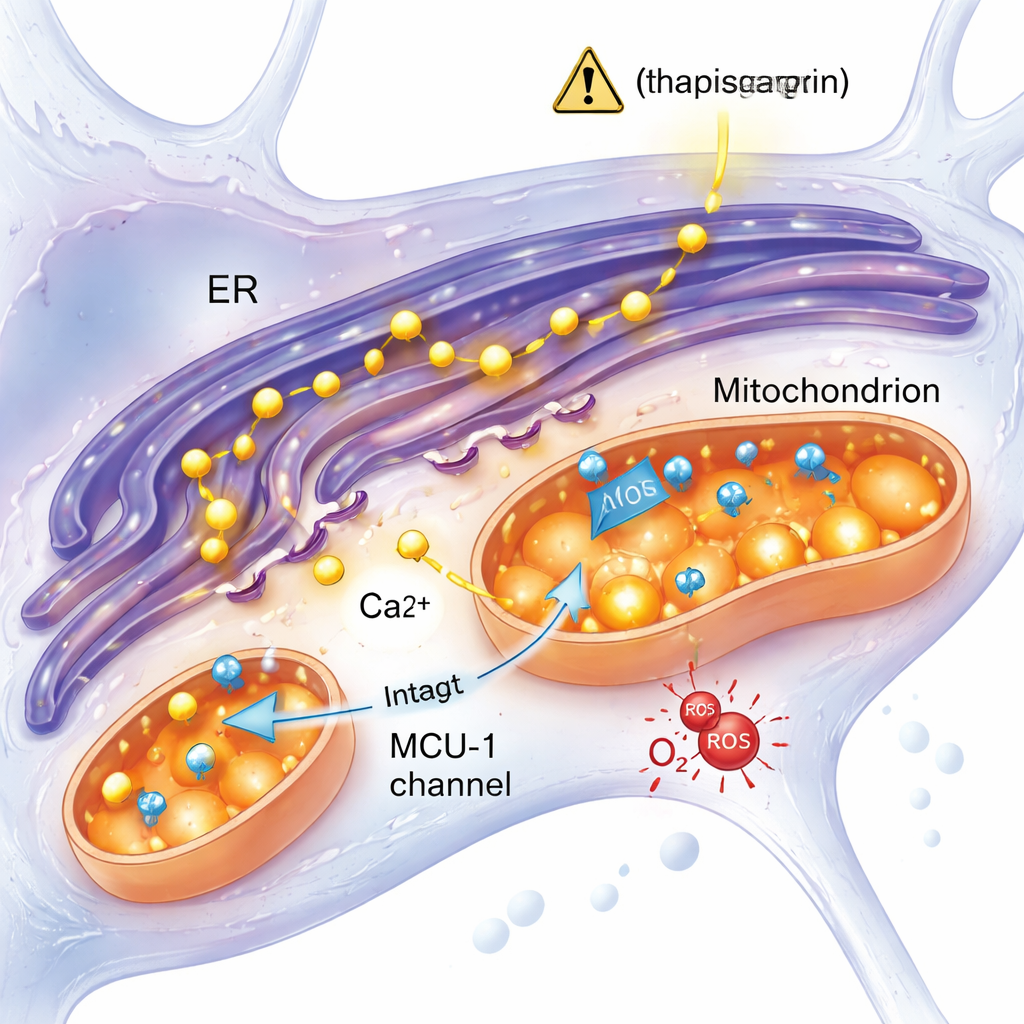
Una risposta allo stress indipendente da un canale chiave per il calcio
Una delle idee principali nella ricerca sull’Alzheimer è che un eccesso di calcio proveniente dal reticolo endoplasmatico si riversi nei mitocondri attraverso un canale chiamato uniportatore del calcio mitocondriale (MCU‑1), sovraccaricandoli e provocandone la disfunzione. Questo studio ha verificato direttamente tale ipotesi. Quando il team ha bloccato MCU‑1 con un inibitore specifico, sia i neuroni di controllo sia quelli mutanti hanno mostrato forti riduzioni nell’assorbimento di calcio mitocondriale, confermando che il canale stesso funzionava in entrambi i gruppi. Inoltre, quando il rilascio di calcio è stato attivato attraverso una via più fisiologica che coinvolge il recettore IP3—un’altra porta chiave per il calcio—le risposte di cellule mutanti e di controllo sono risultate simili. Questi risultati depongono contro l’ipotesi di un MCU‑1 difettoso e suggeriscono invece che siano alterati i contatti fisici e funzionali tra reticolo endoplasmatico e mitocondri, o altri aspetti della loro interazione, nei neuroni portatori della mutazione.
Cosa significa per comprendere e trattare la malattia
Nel complesso, i risultati descrivono neuroni umani portatori della mutazione PS1 M146L come cellule che appaiono normali a riposo ma reagiscono in modo anomalo sotto stress. I loro mitocondri non riescono a captare abbastanza calcio quando i depositi interni vengono rilasciati improvvisamente, eppure funzionano a un livello più elevato—consumando più ossigeno e generando più specie reattive dell’ossigeno—come se fossero bloccati in una modalità compensatoria costosa. Poiché questo avviene in neuroni derivati da esseri umani vivi prima di qualsiasi sintomo clinico, il lavoro sostiene l’idea che la segnalazione del calcio alterata e il sovraccarico mitocondriale precoce siano eventi a monte nell’Alzheimer, non solo prodotti tardivi. Per i non specialisti, il messaggio chiave è che mantenere l’equilibrio tra segnali di calcio e produzione energetica mitocondriale potrebbe essere altrettanto centrale nella prevenzione della malattia quanto mirare alle più note placche amiloidi.
Citazione: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Parole chiave: Malattia di Alzheimer, mitocondri, segnalazione del calcio, mutazione di presenilina‑1, neuroni derivati da iPSC