Clear Sky Science · it
Disgregazione dell’omeostasi del ferro intracellulare tramite disfunzione mitocondriale associata alla soppressione dell’espressione di ATP13A2
Perché il ferro all’interno delle cellule cerebrali è importante
La malattia di Parkinson è più nota per i tremori e la rigidità dei movimenti, ma nel profondo delle cellule cerebrali colpite si svolge un altro dramma: il ferro, un metallo essenziale, comincia ad accumularsi dove non dovrebbe. Questo studio pone una domanda semplice ma fondamentale: come avviene questo accumulo di ferro e in che modo può danneggiare le piccole centrali energetiche e i centri di riciclo all’interno dei neuroni? Rispondendo, il lavoro offre indizi sul perché certe regioni cerebrali degenerano nel Parkinson e nei disturbi correlati, e indica nuove direzioni terapeutiche che vanno oltre la semplice sostituzione della dopamina.
Uno sguardo più da vicino a un raro indizio genetico
I ricercatori si concentrano su una rara forma ereditaria di Parkinson, chiamata PARK9, causata da difetti in un gene denominato ATP13A2. Questo gene codifica per una proteina localizzata nei lisosomi, i compartimenti cellulari deputati allo smaltimento e al riciclo. Le persone con mutazioni in ATP13A2 possono sviluppare anche una condizione caratterizzata da depositi di ferro nel cervello. Questo legame ha reso ATP13A2 un punto di partenza ideale per studiare come l’equilibrio del ferro si alteri. Usando una linea cellulare umana con caratteristiche neuronali che sovraesprime la proteina parkinsoniana alfa‑sinucleina, il gruppo ha impiegato piccoli frammenti di RNA per ridurre l’espressione di ATP13A2 e ha quindi monitorato come cambiassero ferro, produzione di energia e salute cellulare.
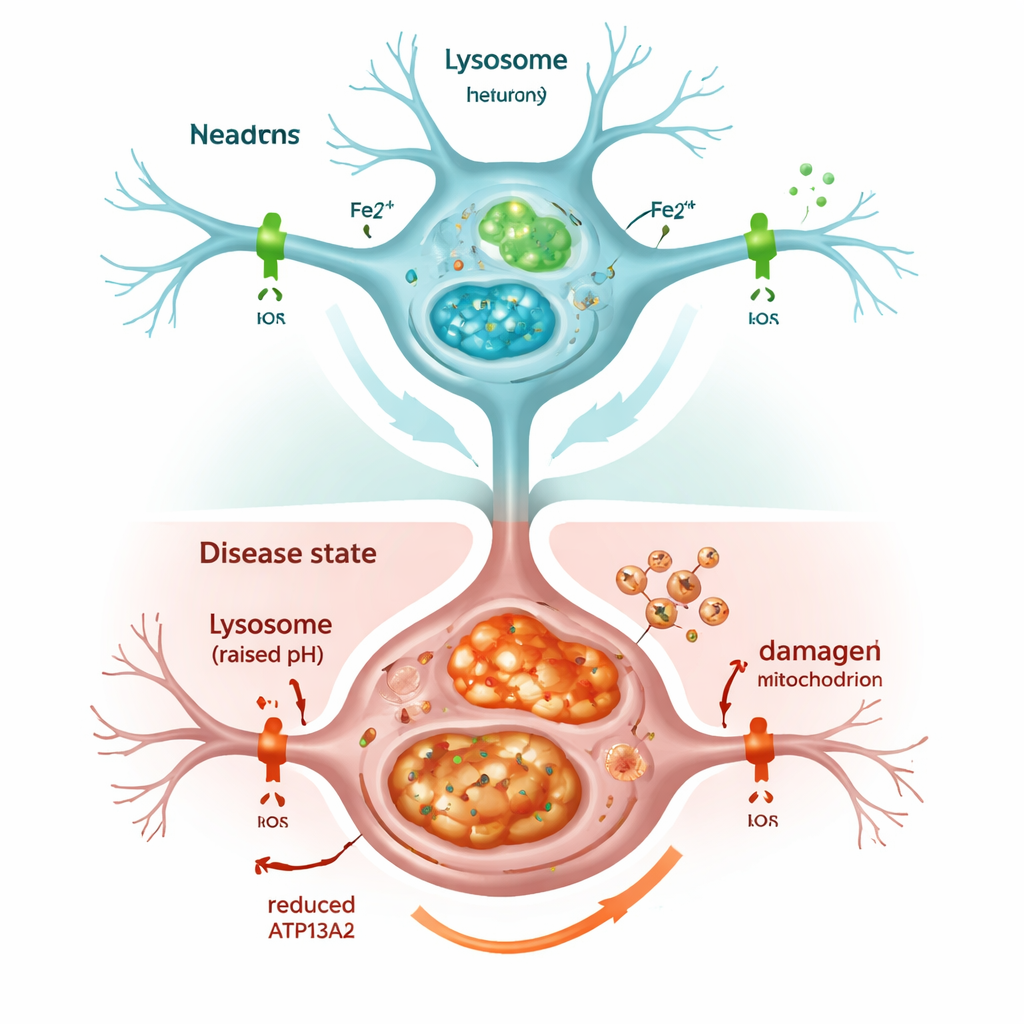
Quando il sistema di riciclo cellulare si inceppa
La soppressione di ATP13A2 ha rapidamente compromesso i lisosomi. Il loro grado di acidità interna, cruciale per degradare materiale indesiderato, è diminuito, e i marcatori del processo di pulizia cellulare, noto come autofagia, si sono accumulati invece di essere eliminati. Di conseguenza si è osservata un’accumulazione di alfa‑sinucleina, in linea con quanto riscontrato nei cervelli dei malati di Parkinson. Le cellule hanno mostrato anche un aumento complessivo del ferro, e in particolare della forma chimicamente attiva, chiamata Fe2+, all’interno sia dei lisosomi sia dei mitocondri. La risposta cellulare è stata un aumento della sintesi di ferritina, la proteina che immagazzina il ferro, ma ciò non è bastato a prevenire i danni: i mitocondri sovraccarichi hanno prodotto un eccesso di specie reattive dell’ossigeno e la sopravvivenza cellulare è diminuita. Il trattamento delle cellule con un farmaco che lega il ferro, simile ad alcuni usati in clinica, ha ridotto questo stress ossidativo e parzialmente salvato la viabilità cellulare, sottolineando che l’eccesso di ferro è stato un fattore chiave del danno.
I sensori del ferro smettono di ascoltare il metallo
Normalmente, le cellule dispongono di un sistema di retroazione che rileva l’aumento del ferro e risponde riducendo l’importazione di ferro. Una proteina chiamata IRP2 percepisce il ferro, in parte tramite un segnale dipendente dall’eme proveniente dai mitocondri, e regola la produzione di proteine trasportatrici del ferro sulla superficie cellulare. Nelle cellule carenti di ATP13A2 questa salvaguardia è venuta meno. I trasportatori che importano ferro nella cellula sono rimasti elevati nonostante i livelli di ferro fossero già aumentati. I livelli della proteina IRP2 sono variati di poco e l’aggiunta di ferro dall’esterno non ha innescato il suo normale degrado. Il gruppo ha ricondotto questo malfunzionamento ai mitocondri: i mitocondri danneggiati respiravano meno efficacemente, presentavano segni di controllo della qualità difettoso (mitofagia alterata) e, cosa cruciale, avevano perso la capacità di sintetizzare l’eme, la molecola contenente ferro che aiuta IRP2 a percepire il ferro. Senza sufficiente eme, IRP2 non riceveva il messaggio di “troppo ferro” e permetteva un continuo afflusso di ferro.
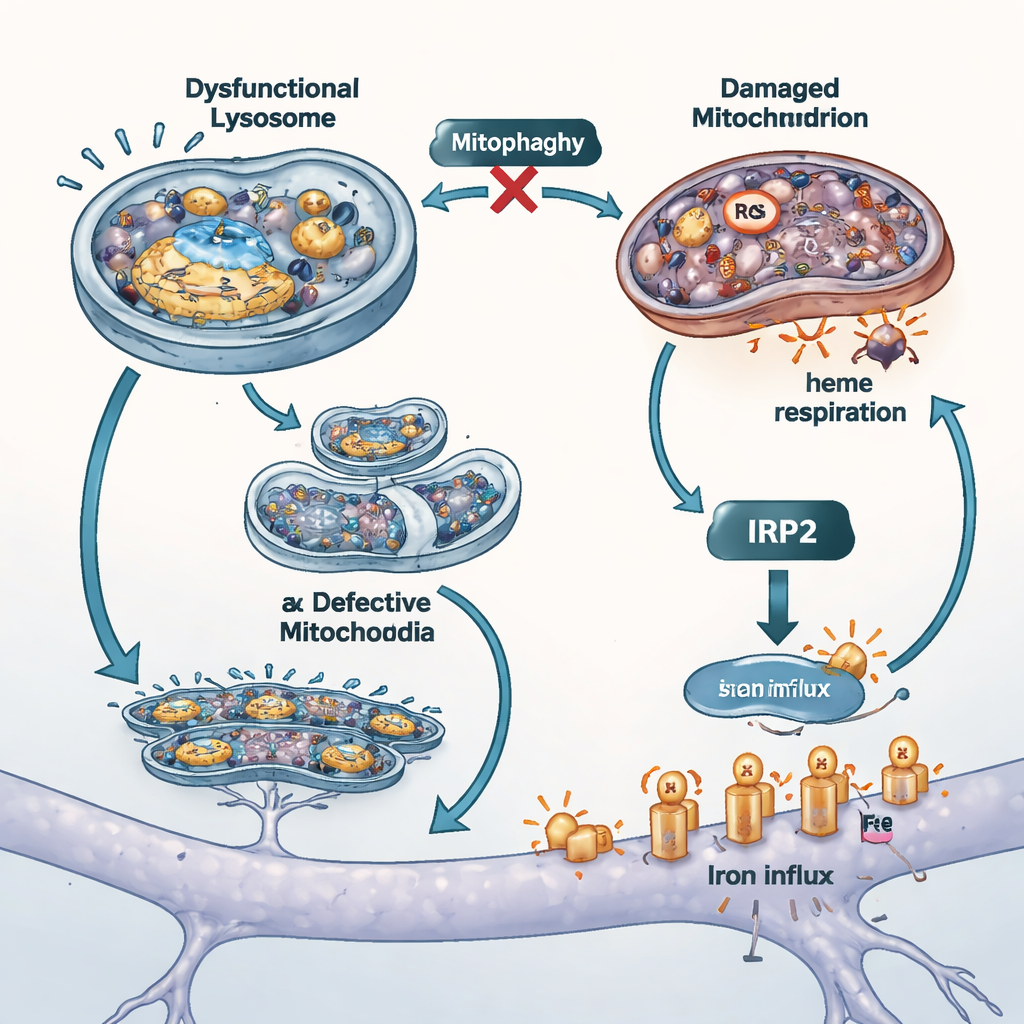
Chiudere il rubinetto del ferro e testare altri modelli
Per valutare quanto questo ingresso incontrollato di ferro contribuisse al danno cellulare, gli scienziati hanno bloccato due principali vie di captazione del ferro. Hanno usato una versione priva di ferro della proteina plasmatica transferrina per competere su un importatore, e un piccolo farmaco per attenuare l’attività di un altro trasportatore chiamato DMT1. Entrambe le manovre hanno ridotto il ferro totale e libero all’interno delle cellule, diminuito lo stress ossidativo mitocondriale e migliorato la sopravvivenza, suggerendo che i canali di membrana per il ferro amplificano il danno quando ATP13A2 viene perso. I ricercatori hanno inoltre ripetuto esperimenti chiave in cellule prive di un altro gene legato al Parkinson, PINK1, noto per compromettere la mitofagia. Queste cellule hanno mostrato la stessa combinazione di accumulo di ferro e ridotta produzione di eme, a sostegno dell’idea che il controllo di qualità mitocondriale e l’equilibrio del ferro siano strettamente intrecciati in diverse forme della malattia.
Cosa significa per il Parkinson e per le terapie future
In termini semplici, lo studio delinea un circolo vizioso. Quando ATP13A2 è soppressa, i lisosomi non riescono a eliminare componenti danneggiati, compresi i mitocondri difettosi. Questi mitocondri indeboliti producono meno energia e meno eme, intaccando il sistema di rilevamento del ferro della cellula. Il ferro continua a entrare attraverso i trasportatori di superficie, si accumula in compartimenti vulnerabili e alimenta reazioni tossiche che danneggiano ulteriormente i mitocondri. Nel tempo, questo circuito può contribuire a spiegare perché alcuni neuroni muoiono nel Parkinson e nei disturbi cerebrali con accumulo di ferro. I risultati suggeriscono che future terapie potrebbero non solo mirare a rimuovere il ferro in eccesso, ma anche a ripristinare la funzione lisosomiale, il controllo di qualità mitocondriale e la produzione di eme—agendo alla fonte del problema piuttosto che limitarsi a rimuovere il metallo dopo che il danno è già avvenuto.
Citazione: Murakami, T., Ohuchi, K., Kiuchi, M. et al. Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression. Sci Rep 16, 5007 (2026). https://doi.org/10.1038/s41598-026-35368-x
Parole chiave: Malattia di Parkinson, ferro cerebrale, mitocondri, lisosomi, sintesi dell’eme