Clear Sky Science · it
Valutazione della base biochimica della resistenza contro l’amiloidosi sistemica
Quando piccole modifiche proteiche bloccano un accumulo letale
Molte malattie infiammatorie croniche, dall’artrite reumatoide alla tubercolosi, possono scatenare una complicanza rara ma spesso fatale chiamata amiloidosi sistemica AA. In questa condizione, una proteina ematica normale si accumula sotto forma di fibre rigide che ostruiscono gli organi. Questo studio pone una domanda sorprendentemente incoraggiante: possono piccole variazioni naturali in quella proteina rendere alcuni animali in larga parte immuni alla malattia — e in tal caso, come?
La minaccia nascosta degli ammassi proteici
L’amiloidosi AA comincia con un segnale infiammatorio nel sangue chiamato serum amyloid A (SAA). Durante infiammazioni severe o prolungate, i livelli di SAA possono aumentare di migliaia di volte rispetto al normale. In alcune persone e animali, una parte di questa proteina si ripiega in modo anomalo e si impila in lunghe fibre, note come fibrille amiloidi, che si diffondono in organi come la milza e i reni. Col tempo queste fibre compromettono la funzionalità degli organi. Eppure non tutti gli individui con alti livelli di SAA sviluppano l’amiloidosi, e alcuni ceppi di topi restano sorprendentemente resistenti anche quando in laboratorio sono spinti verso la malattia. Capire il perché potrebbe indicare nuove strategie per prevenire l’accumulo amiloide nell’uomo.
Topi resistenti e le loro versioni proteiche speciali
Nei topi, la maggior parte delle fibrille amiloidi AA proviene da una versione di SAA chiamata SAA1.1, fortemente associata alla malattia. Tuttavia, alcuni ceppi di topi producono prevalentemente versioni leggermente alterate, denominate SAA1.5 e SAA2.2, e questi ceppi raramente sviluppano l’amiloidosi sistemica AA. Le proteine differiscono solo per una manciata di mattoni costitutivi (amminoacidi), eppure queste differenze si concentrano in una regione strettamente impaccata che forma il nucleo interno delle fibrille patogene. I ricercatori hanno proposto che queste piccole differenze non impediscano del tutto l’aggregazione delle proteine, ma piuttosto le ostacolino dall’assumere la forma fibrillare specifica che è dannosa.
Mettere alla prova le proteine in laboratorio
Per sondare questa idea, il gruppo ha prodotto in batteri tutte e tre le varianti murine di SAA e ha osservato il loro comportamento in esperimenti in provetta. Hanno monitorato la formazione di fibrille usando un colorante fluorescente che si illumina quando si forma l’amiloide, e hanno verificato le strutture con microscopi elettronici. La SAA1.1 legata alla malattia formava facilmente lunghe fibrille diritte. La SAA2.2 poteva anch’essa formare fibrille, ma erano più spesse, più torsionate e strutturalmente più varie, e non producevano lo stesso forte segnale del colorante. La SAA1.5, al contrario, non riusciva a formare fibrille nelle condizioni testate. Quando gli scienziati aggiunsero minuscoli campioni di fibrille patologiche prelevate dalla milza di topi malati come “semi”, la SAA1.1 crebbe rapidamente nuove fibrille che riproducevano da vicino la struttura dell’originale, proprio come un prione. Colpisce che SAA1.5 e SAA2.2 non si siano affatto aggiunte a questi semi; le fibrille ex vivo non riuscivano a reclutarle nella conformazione patogena.
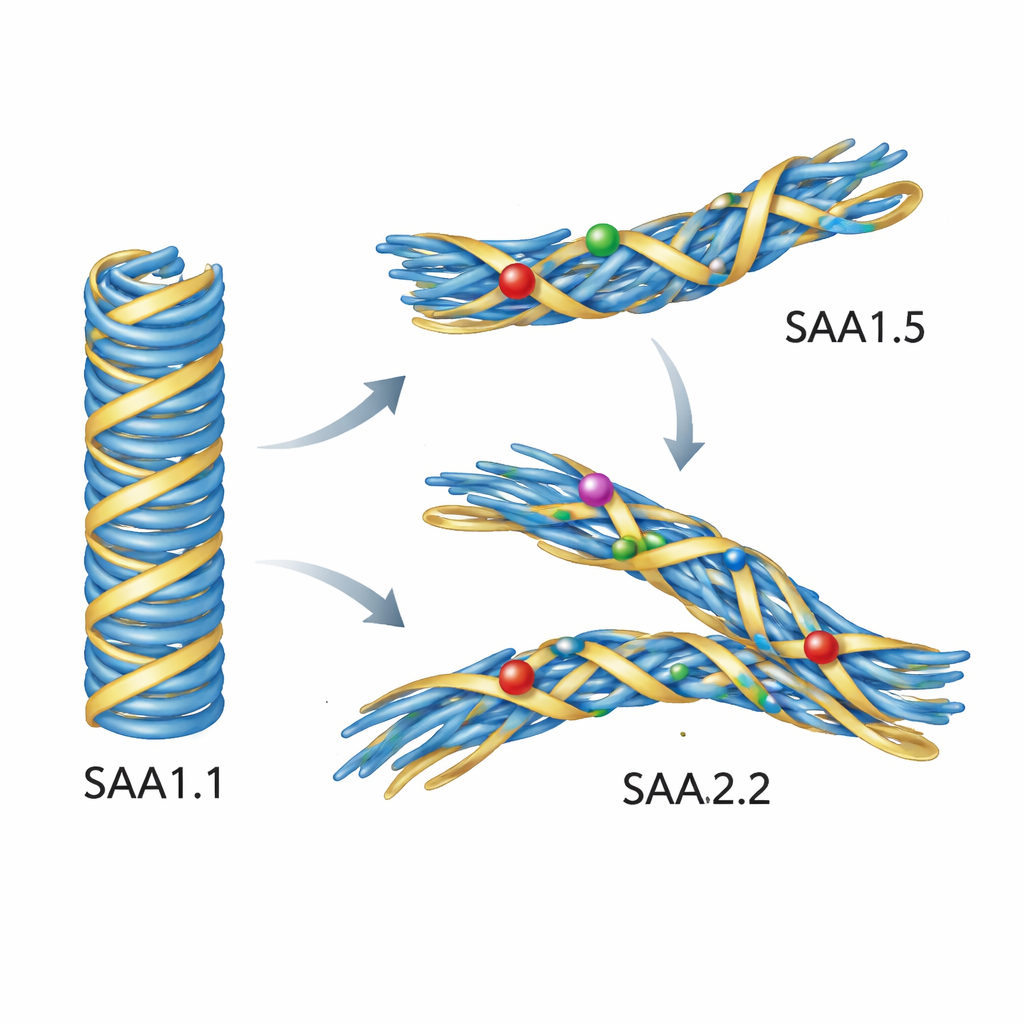
Simulazioni rivelano perché le proteine mutate rifiutano la forma dannosa
I soli esperimenti non potevano mostrare esattamente cosa accade a livello atomico, così gli autori si sono rivolti a simulazioni informatiche dettagliate. Sono partiti da una struttura ad alta risoluzione di una fibrilla AA murina patogena costruita da SAA1.1, quindi hanno sostituito computazionalmente le sequenze con quelle di SAA1.5 e SAA2.2. Simulando queste fibrille in acqua a temperatura corporea, il modello basato su SAA1.1 rimase straordinariamente stabile. Al contrario, le fibrille costruite da SAA1.5 o SAA2.2 si spostarono e si deformarono. Una regione ad ansa chiave nel nucleo si spostò verso l’esterno allentando il contatto con il segmento iniziale della proteina, e diverse catene laterali si riconfigurarono in nuove orientazioni. Questi sottili riarrangiamenti disruppero l’impaccamento compatto che definisce il ripiegamento associato alla malattia. In altre parole, le sequenze varianti non avevano problemi a formare fibrille in generale — ma non riuscivano ad adattarsi comodamente al progetto della fibrilla AA patogena.
Come il progetto della natura suggerisce terapie future
Nel complesso, il lavoro mostra che i ceppi murini “resistenti all’amiloide” non sono protetti perché la loro SAA non possa aggregarsi affatto. Piuttosto, le loro versioni di SAA sono strutturalmente incompatibili con quella forma fibrillare particolare che causa l’amiloidosi sistemica AA. Le proteine possono ancora aggregarsi, ma lo fanno in forme alternative apparentemente benigne. Mutazioni protettive simili sono note in altre malattie da ripiegamento proteico, compresi alcuni casi di prioni e Alzheimer. Questo suggerisce un principio più generale: modificare una proteina incline alla malattia in modo che non possa adottare la sua architettura tossica — pur mantenendo la funzionalità normale — potrebbe essere sufficiente per prevenire la malattia. A lungo termine, terapie ispirate a queste varianti naturali “resistenti”, o a brevi frammenti derivati da esse, potrebbero aiutare a indirizzare le proteine lontano da piegature dannose verso piegature innocue.
Citazione: Moderer, T., Schnell, A.F., Scheurmann, N.J. et al. Assessment of the biochemical basis underlying the resistance against systemic amyloidosis. Sci Rep 16, 1313 (2026). https://doi.org/10.1038/s41598-026-35297-9
Parole chiave: Amiloidosi AA, serum amyloid A, ripiegamento errato delle proteine, resistenza all’amiloide, modelli murini