Clear Sky Science · it
Stochastic LASSO per dati genomici estremamente ad alta dimensionalità
Trovare gli aghi nei pagliai genomici
La biologia moderna può misurare decine di migliaia di geni contemporaneamente, ma gli studi sui pazienti spesso comprendono solo poche centinaia di individui. Nello squilibrio tra queste dimensioni si nascondono piccoli insiemi di geni che contano davvero per prevedere il rischio di malattia o la sopravvivenza. Questo articolo presenta “Stochastic LASSO”, un metodo statistico progettato per individuare in modo affidabile quei geni chiave in oceani di dati genomici rumorosi, anche quando i geni sono molto più numerosi dei pazienti.
Perché scegliere i geni giusti è così difficile
I ricercatori spesso fanno affidamento su strumenti come il LASSO, che riducono gli effetti dei geni non importanti verso zero mantenendo quelli più informativi. Le versioni classiche del LASSO, tuttavia, incontrano difficoltà quando il numero di geni sovrasta quello dei campioni, come avviene comunemente nella genomica dei tumori. Il LASSO standard può selezionare al massimo tanti geni quanti sono i pazienti e tende a trascurare geni che si comportano in modo simile tra loro. Miglioramenti precedenti che introducono penalizzazioni aggiuntive possono gestire in parte questa correlazione, ma rischiano anche di offuscare il significato biologico obbligando geni correlati ad agire come se tutti influenzassero gli esiti nella stessa direzione.
Costruire campioni casuali più puliti
Una soluzione promettente è adattare ripetutamente il LASSO su molti sottoinsiemi più piccoli di geni estratti a caso e poi combinare i risultati. Tuttavia questi approcci di “bootstrap” soffrono ancora di tre problemi: i geni correlati possono annullarsi a vicenda, molti geni vengono raramente o mai campionati e la pura casualità rende la selezione finale instabile. Stochastic LASSO affronta questi problemi con un nuovo schema di campionamento chiamato bootstrap basato sulla correlazione. Invece di scegliere i geni a caso, favorisce intenzionalmente i geni meno correlati con quelli già selezionati, ottenendo insiemi più piccoli di geni molto più indipendenti. Garantisce inoltre che ogni gene venga usato lo stesso numero di volte attraverso le ripetizioni del bootstrap, così nessun gene viene ignorato ingiustamente. 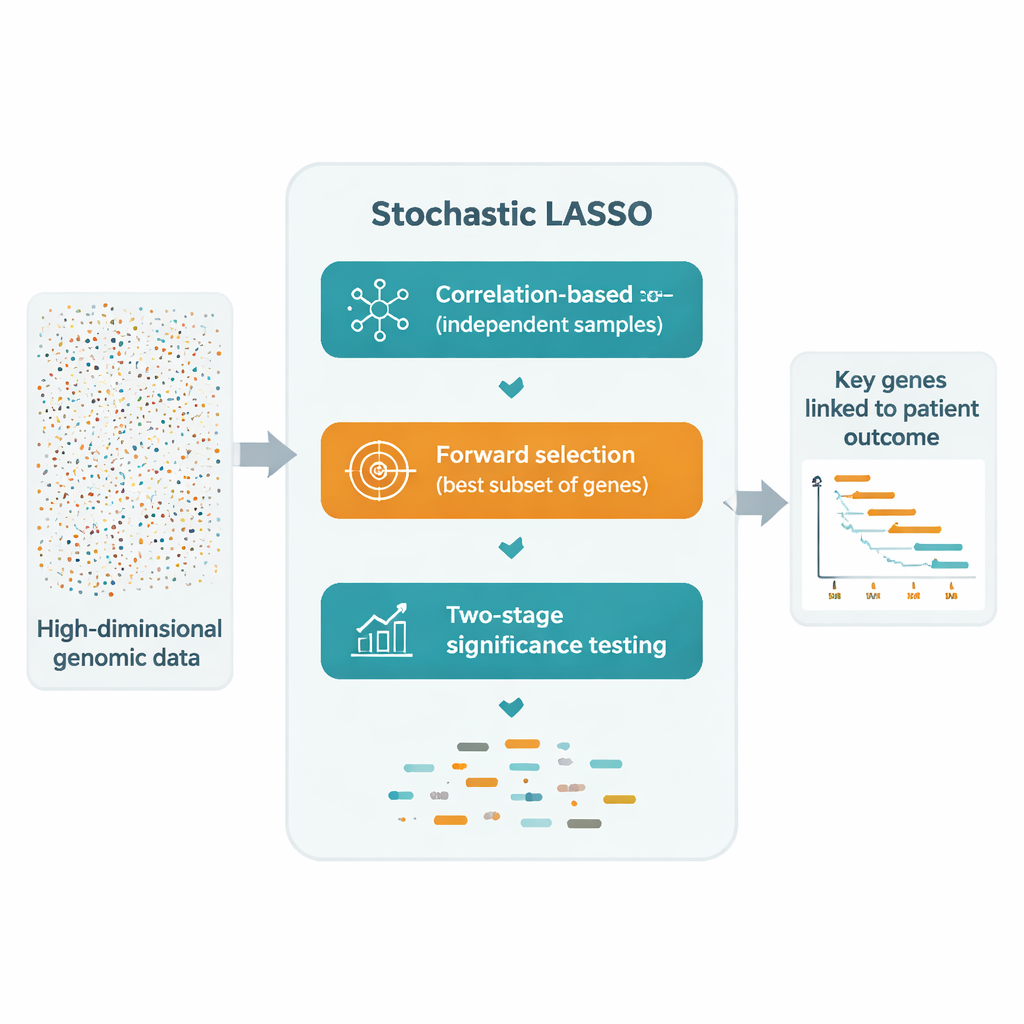
Dai segnali locali a un insieme genico globale
Dopo aver costruito questi sottoinsiemi più puliti, Stochastic LASSO registra quanto è grande il coefficiente di ciascun gene attraverso tutte le stime bootstrap. Questo effetto assoluto medio diventa un “punteggio locale” che riflette quanto costantemente importante appare il gene. Piuttosto che testare esaustivamente ogni possibile combinazione, il metodo costruisce modelli candidati aggiungendo i geni in ordine di punteggio locale e valuta quanto bene ciascun candidato predice gli esiti su dati di validazione separati. In questo modo si arriva a un insieme compatto di geni i cui segnali combinati spiegano nel modo migliore i dati, usando molto meno prove rispetto ai metodi stepwise tradizionali.
Verificare quali geni contano davvero
Per passare da “spesso selezionato” a “statisticamente convincente”, gli autori introducono un test t a due stadi. Innanzitutto verificano se il coefficiente medio di ciascun gene attraverso i bootstrap è chiaramente diverso da zero, segnalandolo come potenzialmente significativo. Poi, tra questi candidati, controllano se l’effetto di ciascun gene è maggiore rispetto alla dimensione d’effetto tipica di tutti i candidati. Solo i geni che superano entrambi i test vengono dichiarati significativi. Poiché questi test si basano sulle numerose stime bootstrap, Stochastic LASSO può identificare con fiducia più geni significativi di quanti siano i pazienti — qualcosa che il LASSO convenzionale non può fare. 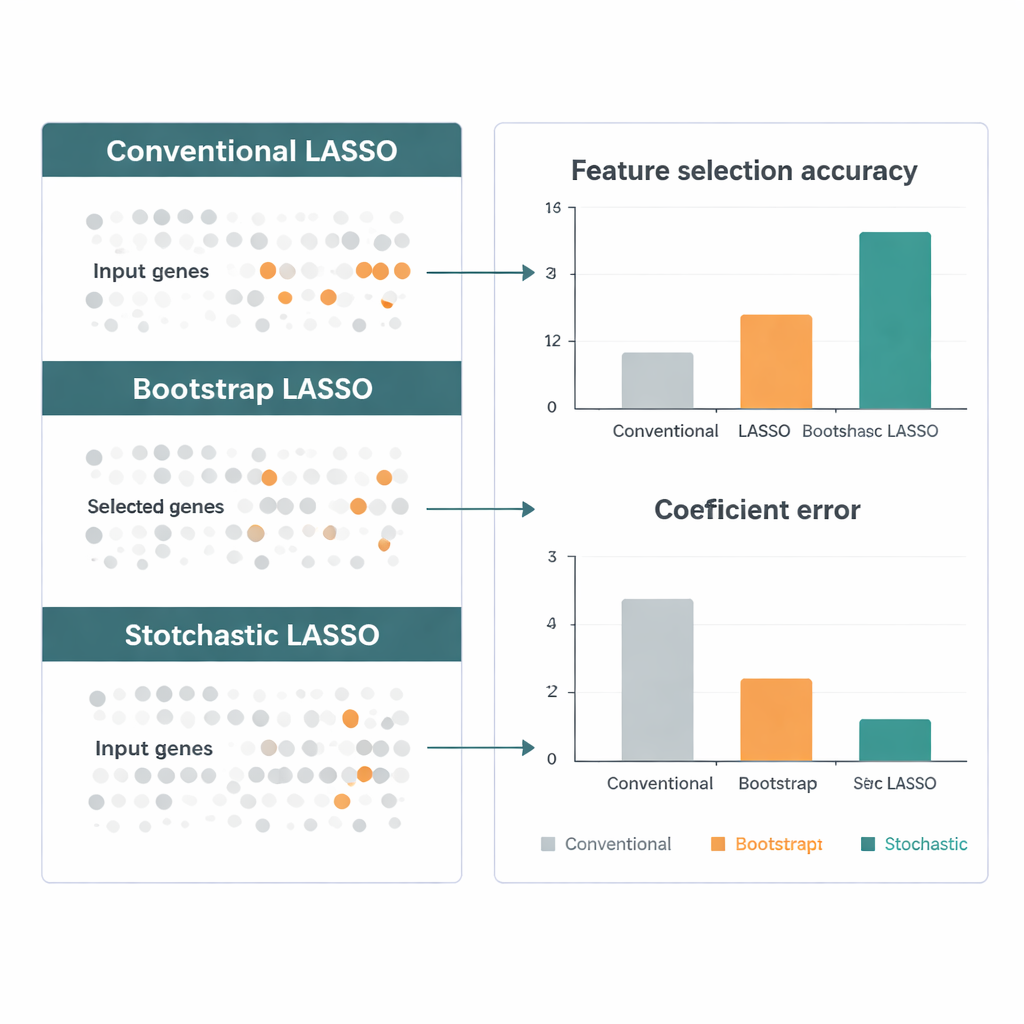
Dimostrare il valore in simulazioni e dati di tumori
Gli autori confrontano Stochastic LASSO con diverse varianti di LASSO di punta usando dati simulati progettati per replicare studi genomici reali: moltissimi geni, forti correlazioni e segnali “veri” noti. In diversi scenari, il nuovo metodo individua i geni corretti più spesso, stima i loro effetti con maggiore accuratezza e rimane stabile da esecuzione a esecuzione. Successivamente applicano il metodo ai dati di espressione genica del The Cancer Genome Atlas sui tumori cerebrali, inclusi gli aggressivi glioblastomi. Stochastic LASSO mette in evidenza centinaia di geni la cui attività è correlata alla sopravvivenza dei pazienti e segnala vie biologiche — come segnali cellulari e percorsi di metabolismo dei farmaci — che hanno supporto indipendente in letteratura, suggerendo che il metodo non è solo statisticamente più efficace ma anche biologicamente sensato.
Che cosa significa per pazienti e ricercatori
Per i non specialisti, il messaggio chiave è che Stochastic LASSO è un filtro più intelligente per i big data genomici. Aiuta gli scienziati a separare i geni realmente legati alla malattia dal rumore statistico, anche quando i dati sono limitati e i geni altamente interconnessi. Fornendo liste di geni e stime d’effetto più accurate e più stabili, può affinare la ricerca di biomarcatori, bersagli farmacologici e firme prognostiche nel cancro e in altre malattie complesse. Sebbene dimostrato su regressione lineare, lo stesso approccio può essere integrato in modelli di sopravvivenza e problemi di classificazione, ampliando il suo impatto potenziale nella ricerca biomedica.
Citazione: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Parole chiave: selezione di caratteristiche genomiche, dati ad alta dimensionalità, metodi LASSO, espressione genica nei tumori, scoperta di biomarcatori