Clear Sky Science · it
Il lattato regola l’asse YTHDF2-FTH1 per promuovere la ferroptosi dei cardiomiociti e aggravare il danno da ischemia-riperfusione miocardica
Perché i pazienti cardiaci dovrebbero interessarsi a questa chimica
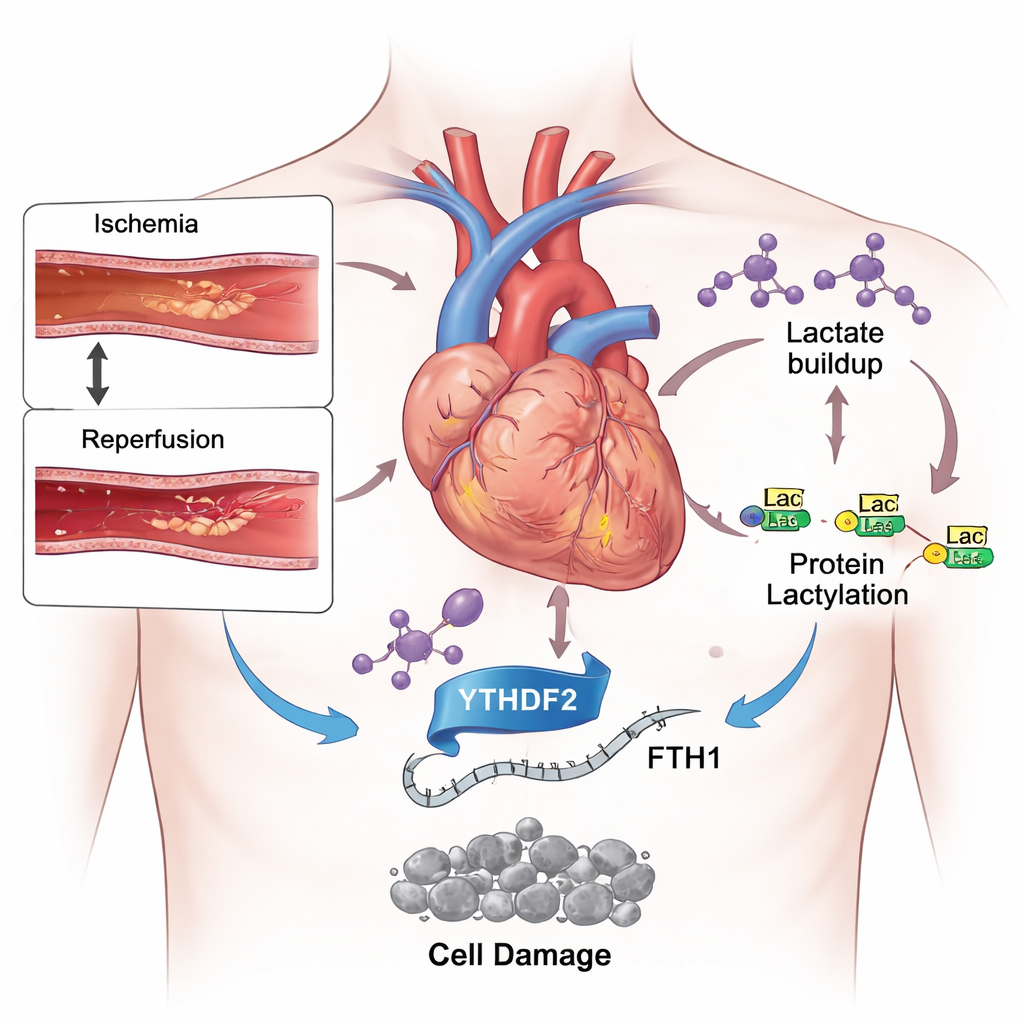
Quando i medici riaprono un’arteria coronaria ostruita dopo un infarto, l’afflusso di sangue fresco salva il muscolo ma può anche provocare danni aggiuntivi, noti come danno da ischemia–riperfusione. Questo studio individua un colpevole sorprendente all’interno delle cellule cardiache: il comune prodotto metabolico lattato. Gli autori mostrano che il lattato può attivare un interruttore molecolare che spinge le cellule cardiache verso un tipo specifico di morte cellulare dipendente dal ferro, peggiorando il danno. Comprendere questa via nascosta potrebbe indicare nuovi farmaci in grado di proteggere meglio il cuore durante il trattamento d’emergenza.
Una lama a doppio taglio nel trattamento dell’infarto
La medicina moderna è diventata molto efficace nel riaprire rapidamente le arterie coronarie ostruite, limitando il danno iniziale dovuto all’infarto. Tuttavia i pazienti possono comunque perdere vaste aree di muscolo cardiaco dopo il ripristino del flusso sanguigno. Una ragione è che il ritorno improvviso di ossigeno e nutrienti genera una tempesta di stress chimico all’interno delle cellule cardiache. Tra i vari tipi di morte cellulare scatenati in questo contesto, una forma più recente chiamata ferroptosi ha attirato l’attenzione. A differenza di forme più familiari come l’apoptosi, la ferroptosi dipende dal ferro e dall’ossidazione incontrollata dei lipidi nelle membrane cellulari, che può compromettere in modo permanente il cuore.
Come il lattato diventa qualcosa in più del semplice “bruciore muscolare”
Durante un infarto, il muscolo cardiaco privato di ossigeno sposta il suo metabolismo verso la glicolisi, un sistema di riserva che degrada rapidamente lo zucchero ma produce grandi quantità di lattato. Utilizzando topi sottoposti a breve occlusione e riapertura di un’arteria cardiaca e cellule cardiache in coltura esposte a ipossia seguita da riperfusione, i ricercatori hanno riscontrato livelli di lattato nettamente aumentati. Allo stesso tempo hanno rilevato un incremento della lattilazione su molte proteine e sugli istoni, gli impalcature che organizzano il DNA. Quando hanno somministrato agli animali un farmaco che rallenta la glicolisi e riduce la produzione di lattato, il danno cardiaco si è ridotto, i marcatori ematici dell’infortunio sono diminuiti e l’equilibrio tra ferro dannoso e antiossidanti protettivi è migliorato. Questi risultati suggeriscono che l’eccesso di lattato non è solo un prodotto di scarto dello stress, ma un motore attivo del danno.
Un interruttore molecolare che allenta il controllo del ferro
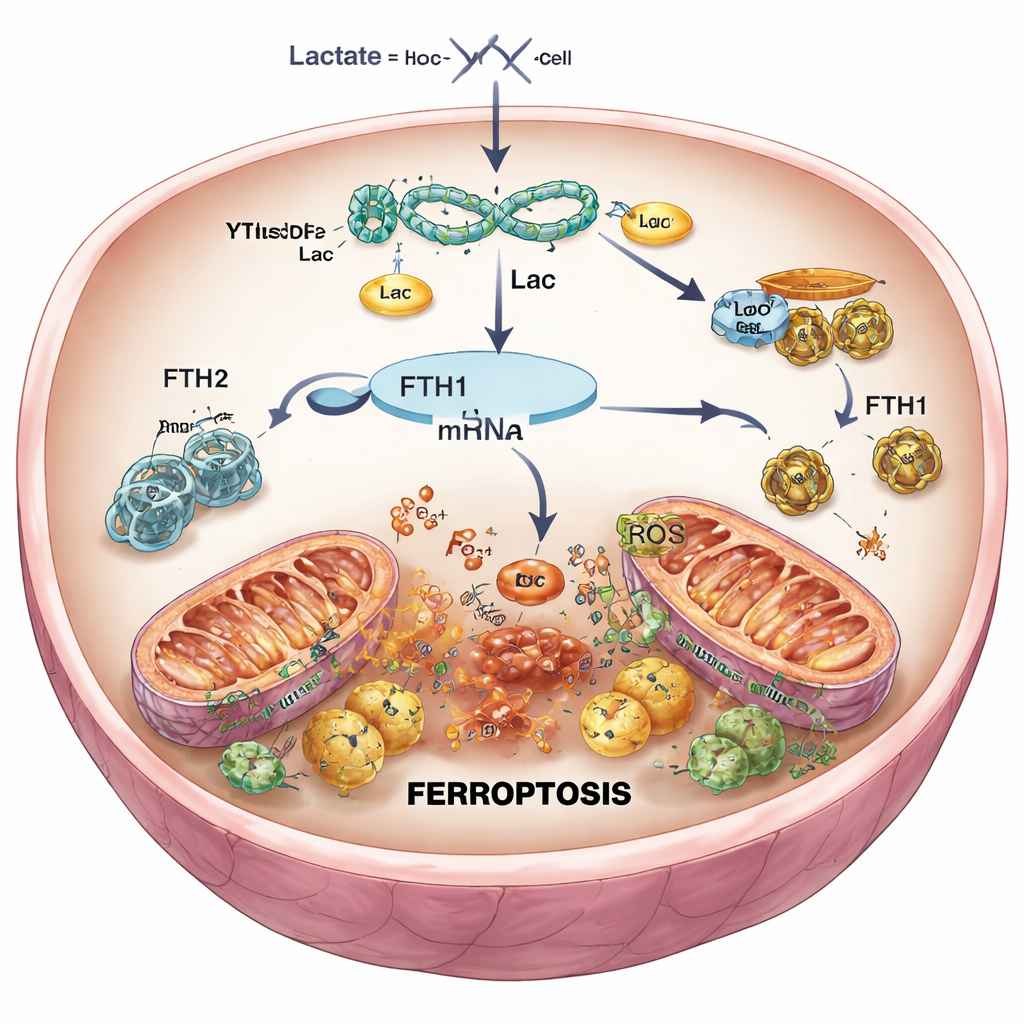
Approfondendo, il team si è concentrato su YTHDF2, una proteina che legge marcature chimiche sull’RNA e decide quanto rapidamente certi messaggi devono essere degradati. Hanno scoperto che ischemia–riperfusione e l’aumento del lattato potenziano entrambi i livelli di YTHDF2 e incrementano la lattilazione attorno al gene che lo codifica, amplificando la sua produzione. Uno degli obiettivi chiave di YTHDF2 si è rivelato essere l’RNA della catena pesante della ferritina 1 (FTH1), una componente centrale della «gabbia» di stoccaggio del ferro nella cellula. FTH1 normalmente sequestra il ferro in una forma sicura, impedendo che alimenti reazioni dannose. Nelle cellule cardiache stressate, YTHDF2 si leava più saldamente all’RNA di FTH1 e ne accelerava il decadimento, lasciando le cellule con meno gabbie di ferritina, più ferro libero, aumento dello stress ossidativo e segni tipici di ferroptosi.
Abbassare il segnale di morte nelle cellule cardiache
Per verificare rapporto di causa ed effetto, i ricercatori hanno usato strumenti genetici per ridurre selettivamente YTHDF2 nelle cellule cardiache e nei topi. Quando YTHDF2 è stato silenziato, i livelli di FTH1 sono risaliti, ferro e specie reattive dell’ossigeno sono diminuiti, i mitocondri hanno mantenuto una morfologia più normale e la sopravvivenza cellulare complessiva è migliorata dopo la riperfusione simulata. Nei topi, meno YTHDF2 significava cicatrici da infarto più piccole e tessuto con aspetto più sano. Tuttavia, quando FTH1 è stato contemporaneamente ridotto, questi benefici sono scomparsi in larga parte: il ferro è risalito, il danno ossidativo è tornato e la dimensione dell’infarto è aumentata. Ciò ha confermato che YTHDF2 promuove la ferroptosi principalmente sopprimendo FTH1, allentando il controllo sul ferro all’interno delle cellule cardiache.
Cosa significa questo per le terapie cardiache future
Combinando i pezzi, lo studio delinea una nuova catena di eventi: un’arteria bloccata e poi riaperta causa accumulo di lattato; il lattato aumenta YTHDF2 tramite lattilazione; YTHDF2 distrugge le istruzioni in RNA per la proteina guardiana del ferro FTH1; e il sovraccarico di ferro risultante innesca la ferroptosi, aggravando il danno cardiaco. Per i pazienti, il messaggio è incoraggiante: questa via offre diversi nuovi punti di intervento. Farmaci che limitano il segnale dannoso del lattato, bloccano la modificazione specifica di YTHDF2 o preservano la funzione di FTH1 potrebbero rendere la riperfusione d’emergenza più sicura e proteggere più tessuto cardiaco. Pur necessitando di conferme nei tessuti umani, questi risultati aprono una via promettente verso trattamenti più delicati ed efficaci per i sopravvissuti all’infarto.
Citazione: Xiang, Z., Xiang, B., Ouyang, T. et al. Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury. Sci Rep 16, 4865 (2026). https://doi.org/10.1038/s41598-026-35130-3
Parole chiave: infarto, lattato, morte cellulare indotta dal ferro, danno da ischemia-riperfusone, protezione dei cardiomiociti