Clear Sky Science · it
Dataset virtuale minimo per un'assemblaggio de novo riproducibile di genomi triploidi
Perché i genomi a tre copie sono importanti
Molte colture e altri organismi non possiedono solo due copie di ogni cromosoma, come gli esseri umani: possono averne tre o più. Ricomporre queste copie extra a partire dai dati di sequenziamento è sorprendentemente difficile, perché le copie sono molto simili ma non perfettamente identiche. Questo articolo presenta un piccolo ma accuratamente progettato dataset “virtuale” che permette ai ricercatori di testare e confrontare i software di assemblaggio genomico su un problema realistico a tre copie (triploide), in condizioni completamente note e riproducibili.
Costruire un genoma sostitutivo semplice
Invece di partire da una pianta o un animale reale, l'autore crea innanzitutto un tratto casuale di DNA lungo un milione di basi per fungere da modello pulito. Questo modello viene quindi duplicato in tre versioni separate, a rappresentare i tre set cromosomici in un organismo triploide. Per imitare come i genomi reali cambiano lentamente nel tempo, lo studio introduce un numero fisso di piccole modifiche—sostituzioni di singole basi—passo dopo passo in ogni copia. Ripetere questo processo per 100 passi produce triplette di genomi che vanno dall'essere quasi identiche a chiaramente ma moderatamente diverse. Questo “gradiente di divergenza” controllato è la spina dorsale del benchmark.
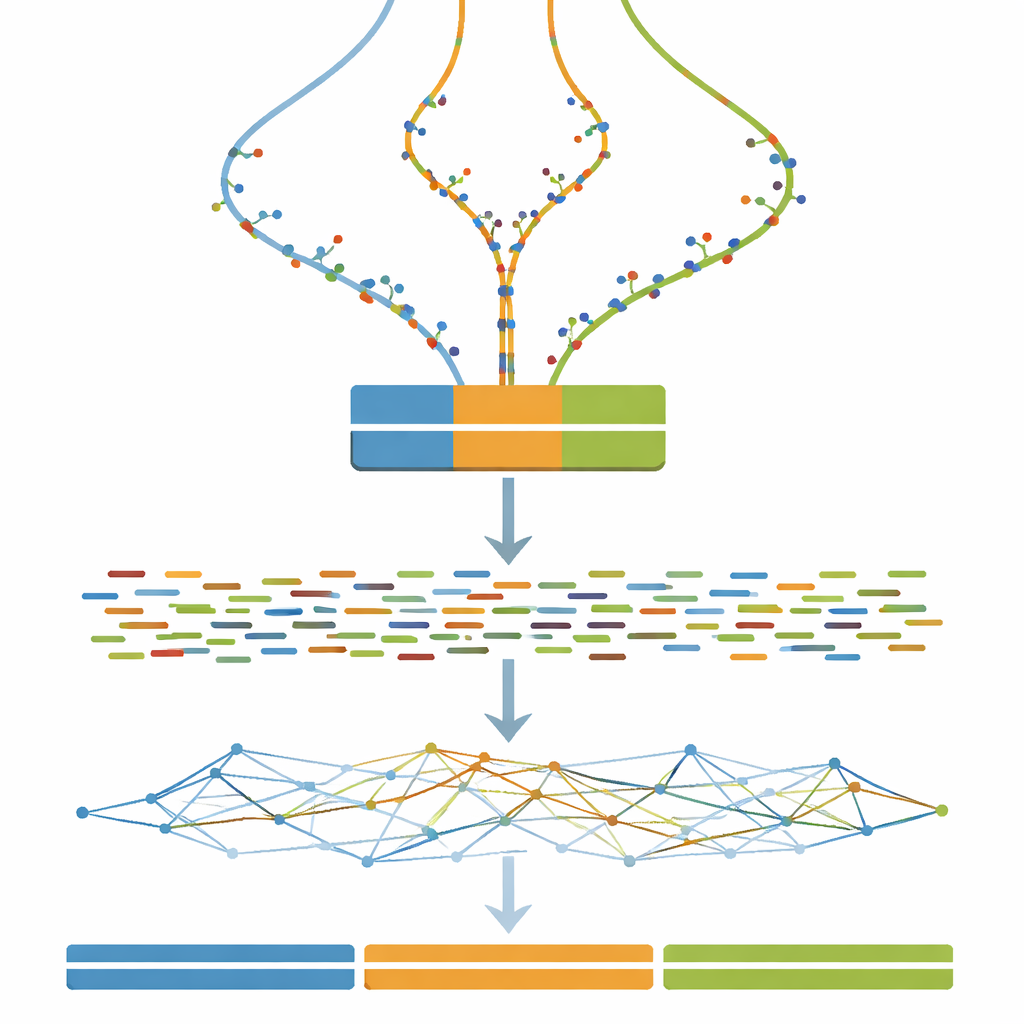
Trasformare i genomi virtuali in esperimenti virtuali
Una volta definito ogni genoma a tre copie, il passo successivo è imitare ciò che vedrebbe una macchina di sequenziamento. Lo studio utilizza software ampiamente adottato per simulare brevi frammenti di DNA accoppiati, simili a quelli prodotti da un sequenziatore Illumina, a profondità di copertura costante e abbastanza elevata. Passaggi di pulizia opzionali imitano pratiche reali comuni, come la correzione di errori casuali di sequenziamento e la fusione di coppie di read sovrapposti. Di conseguenza, chiunque utilizzi il dataset può testare non solo i propri algoritmi di assemblaggio, ma anche come le scelte tipiche di pre-elaborazione influenzano i genomi assemblati finali.
Stress-testare le strategie di assemblaggio
Il cuore del lavoro è un enorme esperimento in cui tutti i read simulati vengono forniti a un singolo programma di assemblaggio genomico cambiando però una sola impostazione chiave: la dimensione del k-mer, un parametro che controlla quanto finemente il software “spezzetta” i read durante la ricostruzione del genoma. Per ogni combinazione di livello di divergenza (da 0 a 100 passi) e dimensione del k-mer (una vasta gamma di valori dispari), viene eseguito un nuovo assemblaggio. Uno strumento di valutazione complementare misura quindi quanto sono continui i pezzi assemblati, quante unità esistono e quanto la loro lunghezza combinata corrisponde alla verità nota di tre milioni di basi. Queste misure vengono riassunte in mappe termiche, rivelando ampie zone dove gli assemblaggi collassano copie diverse in una sola, si frammentano in molti pezzi piccoli o si avvicinano all'ideale di tre contig lunghi e accurati.
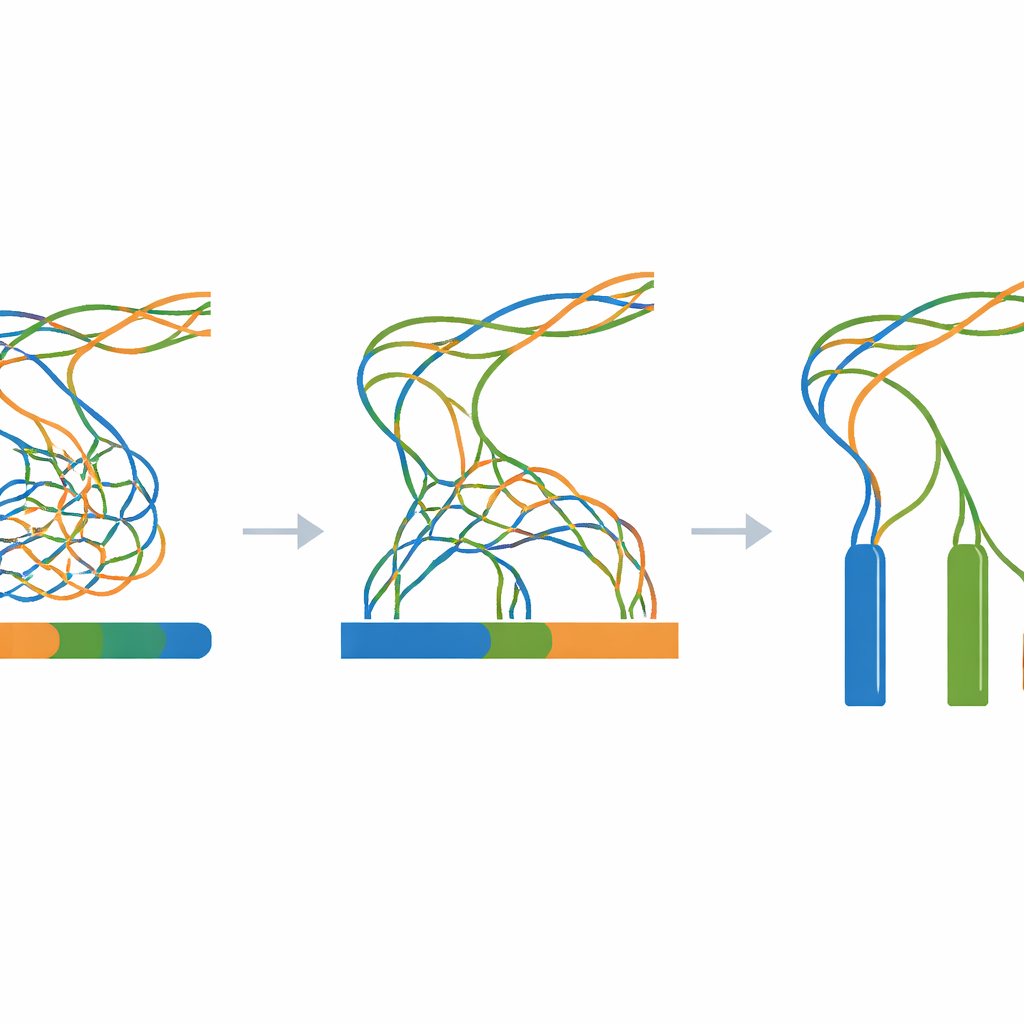
Un riferimento trasparente per genomi complessi
Poiché ogni fase è sintetica e scriptata—from il modello casuale iniziale fino agli assemblaggi finali—i ricercatori possono riprodurre l'intero flusso di lavoro su qualsiasi computer Linux standard utilizzando solo strumenti open-source. L'archivio Zenodo collegato nell'articolo contiene il genoma modello, tutte le sequenze mutate intermedie, tutti i read simulati e ogni risultato di assemblaggio, insieme ai log e a semplici script di supporto. Controlli tecnici confermano che il processo di mutazione si comporta come previsto, che i read simulati corrispondono alle lunghezze e alla copertura richieste e che gli assemblaggi mostrano lo schema previsto: forte over-collapsing quando le tre copie sono quasi identiche e separazione più chiara man mano che si allontanano tra loro.
Cosa significa in parole semplici
In termini quotidiani, questo articolo offre una pista di prova controllata per software che cerca di ricostruire tre libri di istruzioni simili a partire da mucchi di frammenti mescolati. Aumentando gradualmente quanto i tre libri siano diversi e variando sistematicamente un'impostazione chiave nel processo di ricostruzione, il dataset rende semplice vedere quando e come i metodi attuali falliscono o hanno successo. Gli sviluppatori possono usarlo per mettere a punto nuovi algoritmi, mentre gli utilizzatori possono capire meglio quali impostazioni funzionano meglio per i genomi triploidi. Sebbene il DNA sia artificiale, le lezioni che consente—sul collasso, sulla separazione e sull'impatto delle scelte di parametri—sono direttamente rilevanti per gli sforzi reali di decodificare i genomi complessi di molte specie importanti.
Citazione: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Parole chiave: assemblaggio di genomi triploidi, benchmarking di poliploidi, dataset DNA sintetico, assemblaggio de novo, ottimizzazione k-mer