Clear Sky Science · it
Prevenire i cimiteri dei dati proteomici attraverso responsabilità collettiva e coinvolgimento della comunità
Perché i tuoi dati medici non dovrebbero finire in un cimitero digitale
La medicina moderna si basa sempre più su enormi insiemi di dati che descrivono le migliaia di proteine in azione nelle nostre cellule. Questi file vengono spesso condivisi apertamente online, con la promessa che altri scienziati possano verificare i risultati o porre nuove domande senza dover svolgere nuovi esperimenti. Ma se i dati vengono pubblicati in formati confusi, privi di dettagli essenziali o legati a software proprietari, diventano «cimiteri dei dati»: visibili a tutti ma praticamente inutilizzabili. Questo articolo mostra come un corso universitario abbia trasformato gli studenti in detective dei dati per far emergere questo problema nascosto — e suggerisce semplici correzioni che potrebbero rendere i dati condivisi veramente riutilizzabili.
Imparare la scienza ripetendo studi reali
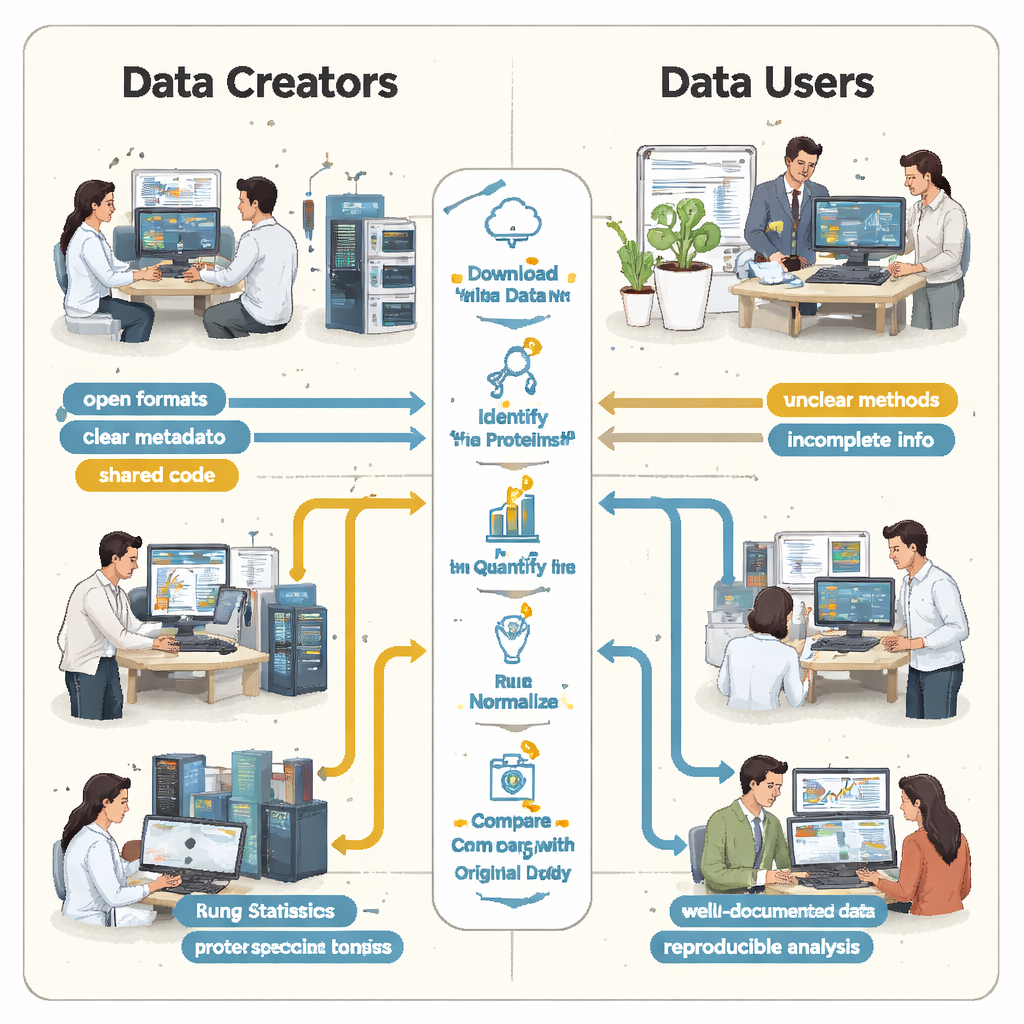
All’Università di Helsinki, agli studenti di laurea magistrale di un corso di proteomica via spettrometria di massa è stato chiesto qualcosa di ambizioso: selezionare dataset proteici reali e pubblici da un grande repository e tentare di riprodurre i risultati pubblicati. Lavorando in piccoli gruppi, hanno scaricato sei progetti dalla rete ProteomeXchange, che ospita risultati di spettrometria di massa provenienti da molti laboratori nel mondo. Utilizzando una pipeline di analisi condivisa in R, gli studenti hanno seguito gli stessi passaggi generali degli autori originali: identificare le proteine, misurarne l’abbondanza, pulire i dati e verificare quali proteine cambiano tra condizioni come tessuto malato e sano.
Grandi promesse, istruzioni mancanti
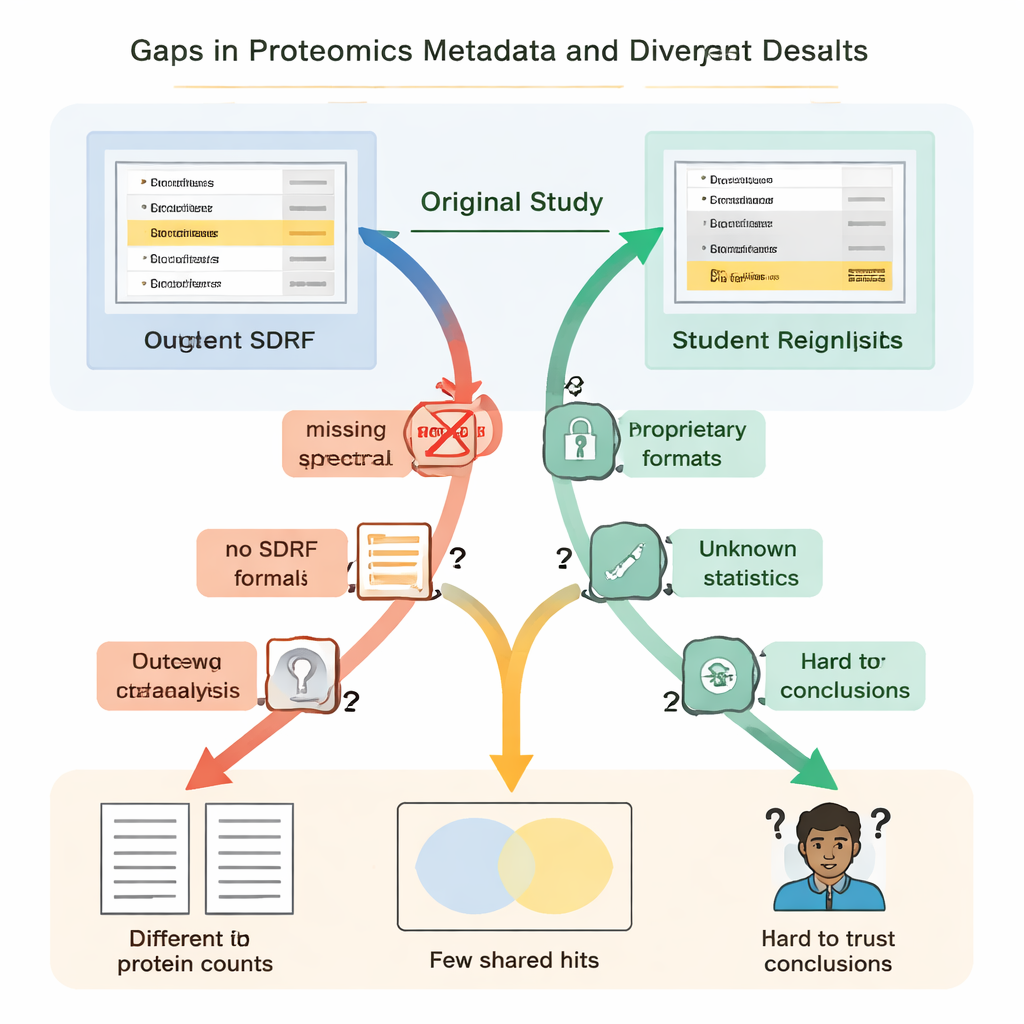
Gli studenti hanno scoperto rapidamente che «aperto» non significava sempre «riutilizzabile». In ogni caso, indicazioni essenziali erano mancanti o difficili da trovare. I collegamenti chiave tra campioni e file di dati non erano descritti in un formato semplice e leggibile dalle macchine, così i gruppi hanno dovuto indovinare quali file raw corrispondessero a quali gruppi biologici leggendo gli articoli e decifrando i nomi dei file. Mancavano dettagli su come fossero controllati i falsi positivi — per esempio l’uso di sequenze proteiche «decoy» — rendendo impossibile valutare in modo rigoroso quanto fossero affidabili le liste di proteine riportate. In diversi progetti, i risultati principali erano bloccati in formati proprietari o dipendevano da software commerciale a cui gli studenti non potevano accedere, costringendoli a rifare ampie parti dell’analisi da zero.
Quando piccole lacune producono grandi differenze
Queste informazioni mancanti non erano solo fastidi; hanno portato a risultati scientifici drasticamente diversi. In uno studio su una malattia renale, gli autori originali riportavano poco meno di cinquemila proteine, mentre la rianalisi degli studenti — usando uno strumento open e una libreria spettrale autocostruita — ne ha trovate oltre tredicimila. Una proteina evidenziata nell’articolo originale come particolarmente importante non compariva in modo convincente nel file di identificazione sottostante e non è stata rilevata affatto nel flusso di lavoro degli studenti. In un altro caso, lo studio originale elencava 108 proteine come differenzialmente espresse tra condizioni, ma gli studenti, partendo dagli stessi dati raw ma senza informazioni complete sulle statistiche originali, sono riusciti a identificare con sicurezza solo 11. Altrove, la mancanza di repliche biologiche nei file caricati rendeva semplicemente impossibile eseguire test statistici adeguati.
Quanto dovrebbe realmente contenere un dataset «riutilizzabile»
Dai sei casi analizzati è emerso un quadro chiaro: le principali barriere alla riproducibilità non erano le macchine di spettrometria di massa in sé, ma il modo in cui i risultati venivano impacchettati e condivisi. Gli autori sostengono che ogni dataset proteomico dovrebbe essere accompagnato da un pacchetto minimo per la rianalisi. Questo include i dati raw insieme a formati di risultato aperti e standard della comunità; una tabella standardizzata che colleghi ciascun campione alle condizioni sperimentali; riepiloghi di controllo di qualità di base; eventuali librerie spettrali o file di sequenze proteiche necessari per ripetere la ricerca; e parametri di analisi e codice completi, idealmente archiviati con contenitori software versionati. Repository, riviste e revisori potrebbero aiutare incoraggiando o richiedendo ai submitter di fornire questo pacchetto fin dall’inizio, in modo che altri non debbano ricostruire il flusso di lavoro a partire da indizi sparsi.
Formare gli scienziati mentre si ripara il sistema
Il corso ha avuto una duplice funzione. Per gli studenti, ha offerto un modo pratico per padroneggiare metodi proteomici complessi, statistica e programmazione, rivelando al contempo quanto le conclusioni pubblicate possano essere fragili quando la documentazione è incompleta. Per la comunità più ampia, le difficoltà degli studenti hanno fornito un test di stress delle pratiche correnti di condivisione dei dati, evidenziando esattamente dove i metadati e i record di analisi sono carenti. Gli autori suggeriscono che corsi simili potrebbero essere organizzati altrove, trasformando le aule in motori di controllo qualità che spingano continuamente verso dati più chiari e trasparenti.
Da cimiteri dei dati a risorse vive
In termini semplici, l’articolo conclude che molti dataset proteici oggi presenti nei repository pubblici rischiano di diventare cimiteri digitali — esperimenti costosi i cui risultati non possono essere verificati o estesi con affidabilità. Tuttavia la soluzione è relativamente semplice: trattare metadati, formati aperti e codice condivisibile come parti integranti dell’esperimento, non come ripensamenti. Se ricercatori, revisori e repository insistono collettivamente su un pacchetto semplice e ben documentato ogni volta che vengono condivisi dati proteomici, quei dataset possono restare «vivi»: pronti per essere rianalizzati, combinati con nuovi studi e utilizzati per rafforzare le evidenze alla base delle scoperte biomediche.
Citazione: Vadadokhau, U., Soliman, M., Castillon, L. et al. Preventing Proteomics Data Tombs Through Collective Responsibility and Community Engagement. Sci Data 13, 287 (2026). https://doi.org/10.1038/s41597-026-06614-8
Parole chiave: proteomica, riproducibilità dei dati, scienza aperta, spettrometria di massa, condivisione dei dati di ricerca