Clear Sky Science · it
AF2BIND: previsione dei siti di legame per piccole molecole usando la rappresentazione a coppie di AlphaFold2
Trovare bersagli farmacologici in un mare di proteine
I medicinali moderni spesso agiscono agganciandosi a piccole nicchie e rientranze sulla superficie delle proteine nelle nostre cellule. Eppure, anche con gli enormi cataloghi di strutture proteiche disponibili oggi, rimane sorprendentemente difficile prevedere in anticipo dove una piccola molecola — un potenziale farmaco — possa effettivamente aderire. Questo studio presenta AF2BIND, uno strumento computazionale semplice ma potente che sfrutta i meccanismi interni di AlphaFold2, il predittore di strutture proteiche di riferimento, per mettere in luce i probabili siti di legame per farmaci su migliaia di proteine umane. L’obiettivo è restringere la ricerca di nuovi medicinali e rivelare punti funzionali nascosti che i metodi tradizionali trascurano.
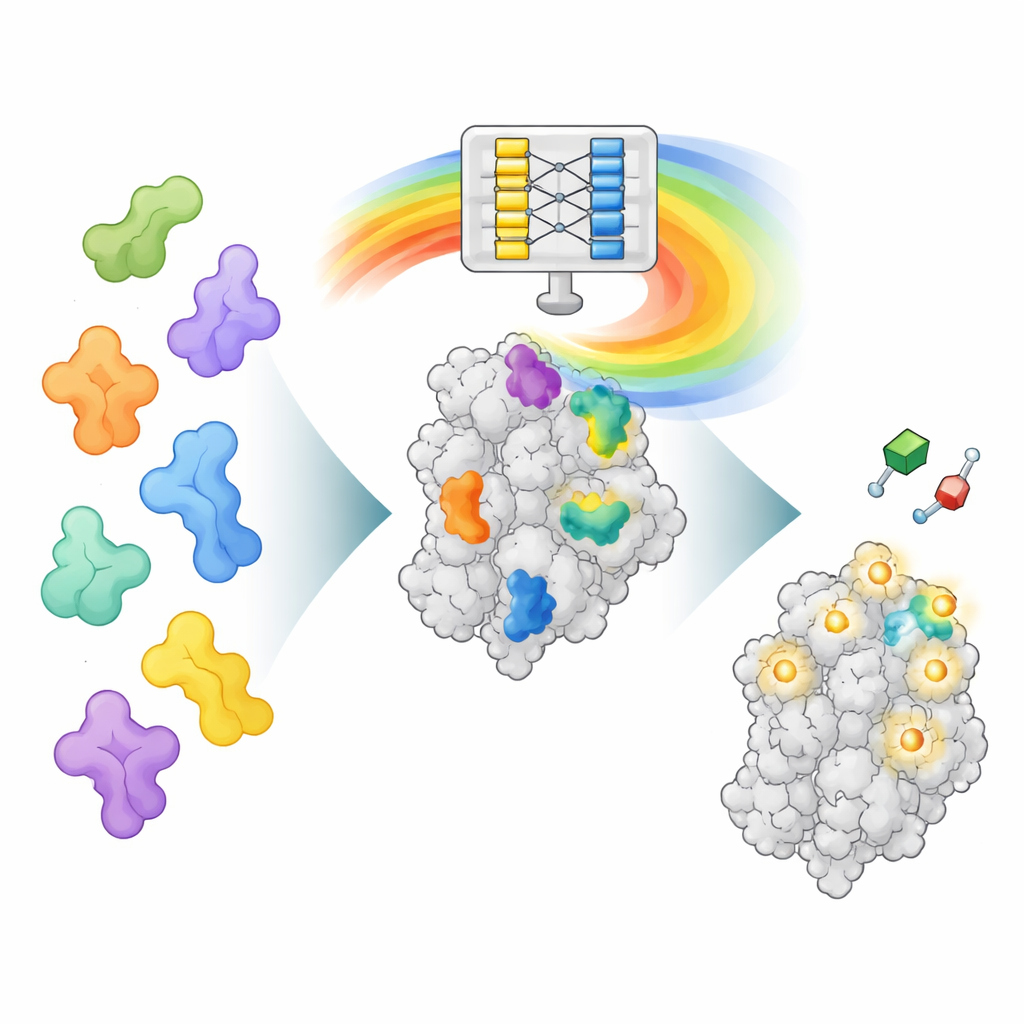
Un nuovo modo di leggere la “mente” di AlphaFold
AlphaFold2 è stato addestrato a prevedere come una catena di amminoacidi si ripiega in una struttura tridimensionale, non a trovare i siti dove si legano i farmaci. Tuttavia, nel processo di apprendimento del ripiegamento, ha acquisito anche ricchi schemi su come diverse parti della proteina interagiscono tra loro. AF2BIND attinge a uno di questi strati dati interni, chiamato rappresentazione a coppie, che codifica come ogni coppia di posizioni amminoacidiche si relaziona nello spazio. Gli autori forniscono ad AlphaFold2 la sequenza proteica insieme alla sua struttura del backbone e aggiungono anche 20 amminoacidi supplementari, uno per ogni tipo, come catene “esca” separate. AlphaFold2 calcola quindi come la proteina interagisce con ciascuna di queste residenze esca. Questi pattern di interazione diventano l’input per un modello di regressione logistica molto semplice che stima, per ogni posizione nella proteina, la probabilità che essa appartenga a un sito di legame per piccole molecole.
Trasformare segnali nascosti in previsioni pratiche
L’addestramento di AF2BIND ha richiesto un set accuratamente curato di circa 1.900 strutture proteina–ligando in cui le piccole molecole erano legate con evidenze sperimentali di alta qualità. I ricercatori si sono prodigati per evitare qualsiasi “imbroglio” dovuto a similarità: hanno suddiviso i dati in modo che le proteine del test non condividessero ripiegamento complessivo, sequenza o neppure la forma della tasca di legame con quelle usate per l’addestramento. In questo rigoroso benchmark, la rappresentazione a coppie di AF2 ha superato diverse alternative di embedding basate su reti neurali, incluse quelle fondate soltanto sulla sequenza o sulla progettazione di sequenze condizionata alla struttura. Utilizzando solo le caratteristiche a coppie, AF2BIND ha recuperato circa due terzi dei residui di legame noti nelle predizioni con punteggio più alto e ha mostrato prestazioni solide sulle metriche di classificazione standard, rimanendo inoltre robusto a modeste variazioni nella forma della proteina e nelle orientazioni delle catene laterali.
Leggere indizi chimici dalle residenze esca
Poiché AF2BIND è un modello lineare semplice, le sue decisioni sono sorprendentemente trasparenti per un sistema di IA moderno. Ognuno dei 20 amminoacidi esca contribuisce con una quantità misurabile al punteggio finale di legame in una data posizione proteica. Esaminando questi contributi su circa 2.000 complessi proteina–ligando, gli autori hanno scoperto che certe combinazioni di esche si attivano più intensamente per ligandi oleosi e ricchi di carbonio, mentre altre si illuminano per molecole più polari e idrofile. In altre parole, il modello di attivazione delle esche agisce come un’impronta chimica grezza del tipo di piccole molecole preferite da una specifica tasca. Ciò suggerisce che in futuro approcci simili ad AF2BIND potrebbero non solo indicare dove un farmaco potrebbe legarsi, ma anche suggerire la chimica che più si adatterebbe a quel sito.
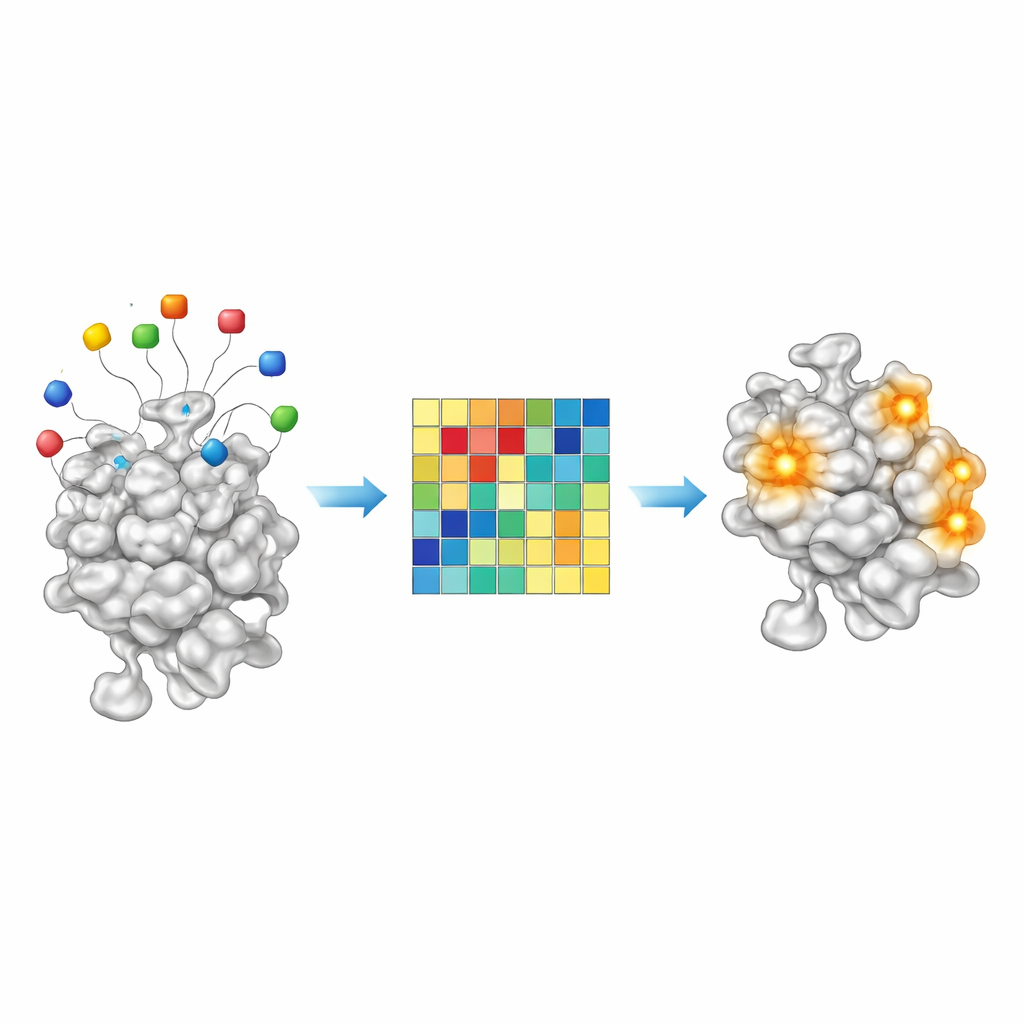
Scansionare il proteoma umano per nuove tasche
Dotata del modello addestrato, la squadra ha quindi applicato AF2BIND alle strutture previste da AlphaFold per l’intero proteoma umano. Dopo aver rimosso regioni a bassa confidenza e suddiviso proteine molto grandi in frammenti strutturali gestibili, hanno raggruppato residui vicini con punteggi elevati in siti di legame candidati. AF2BIND ha previsto oltre 20.000 siti di questo tipo in più di 13.000 proteine. Colpisce il fatto che la maggior parte di questi non sovrapponesse le tasche individuate da metodi basati su omologia come AlphaFill, che copia ligandi da strutture cristalline correlate, né con un diffuso cercatore di tasche chiamato P2Rank. Molti siti individuati solo da AF2BIND sono meno profondi o più diffusi rispetto alle tasche classiche sepolte e spesso coincidono con regioni che legano peptidi, RNA, DNA o altre proteine — interfaccie che tuttavia potrebbero essere raggiungibili da piccole molecole.
Implicazioni per la scoperta di farmaci e le malattie
Per valutare quanto questi nuovi siti suggeriti possano essere promettenti per il design di farmaci, gli autori hanno usato uno strumento indipendente che valuta la “druggabilità” basandosi su dimensione della tasca, chiusura ed ambiente chimico. In media, i siti individuati da AF2BIND hanno ottenuto punteggi superiori a una soglia comune per bersagli farmacologici interessanti, inclusi quelli presenti in proteine associate a malattie ereditarie. Incrociando i risultati con esperimenti chemoproteomici che mettono in luce cisteine reattive nelle cellule, AF2BIND e P2Rank insieme hanno spiegato quasi la metà delle regioni osservate come ligabili, con ciascun metodo capace di cogliere casi sfuggiti all’altro. Il lavoro dimostra che le rappresentazioni interne apprese dalle reti di predizione strutturale possono essere riutilizzate per mappare a grande scala i probabili siti di legame per farmaci, senza conoscenza preliminare di alcun ligando specifico. Per i non specialisti, il messaggio chiave è che le stesse svolte tecnologiche dell’IA che prevedono le forme delle proteine stanno cominciando a rivelare dove e come i farmaci potrebbero agganciarsi a quelle forme, accelerando potenzialmente la ricerca di nuovi trattamenti e illuminando punti di controllo precedentemente nascosti nelle nostre proteine.
Citazione: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Parole chiave: siti di legame delle proteine, scoperta di farmaci, AlphaFold2, biologia computazionale, bioinformatica strutturale