Clear Sky Science · it
DECODE: framework comune di deconvoluzione basato sul deep learning per diversi dati omici
Perché questa ricerca è importante
La biomedicina moderna è sommersa da misurazioni dei nostri tessuti: quali geni sono attivi, quali proteine sono presenti e quali piccole molecole alimentano le nostre cellule. Tuttavia la maggior parte di queste misurazioni viene effettuata su campioni miscelati, in cui molti tipi cellulari sono mescolati insieme. Lo studio che presenta DECODE introduce un potente framework di intelligenza artificiale in grado di separare questi segnali, indicando quali cellule e stati cellulari sono presenti, anche attraverso tipi di dati molto diversi. Questa capacità potrebbe accelerare la ricerca su cancro, immunità e malattie metaboliche sfruttando meglio i campioni già presenti nelle biobanche.
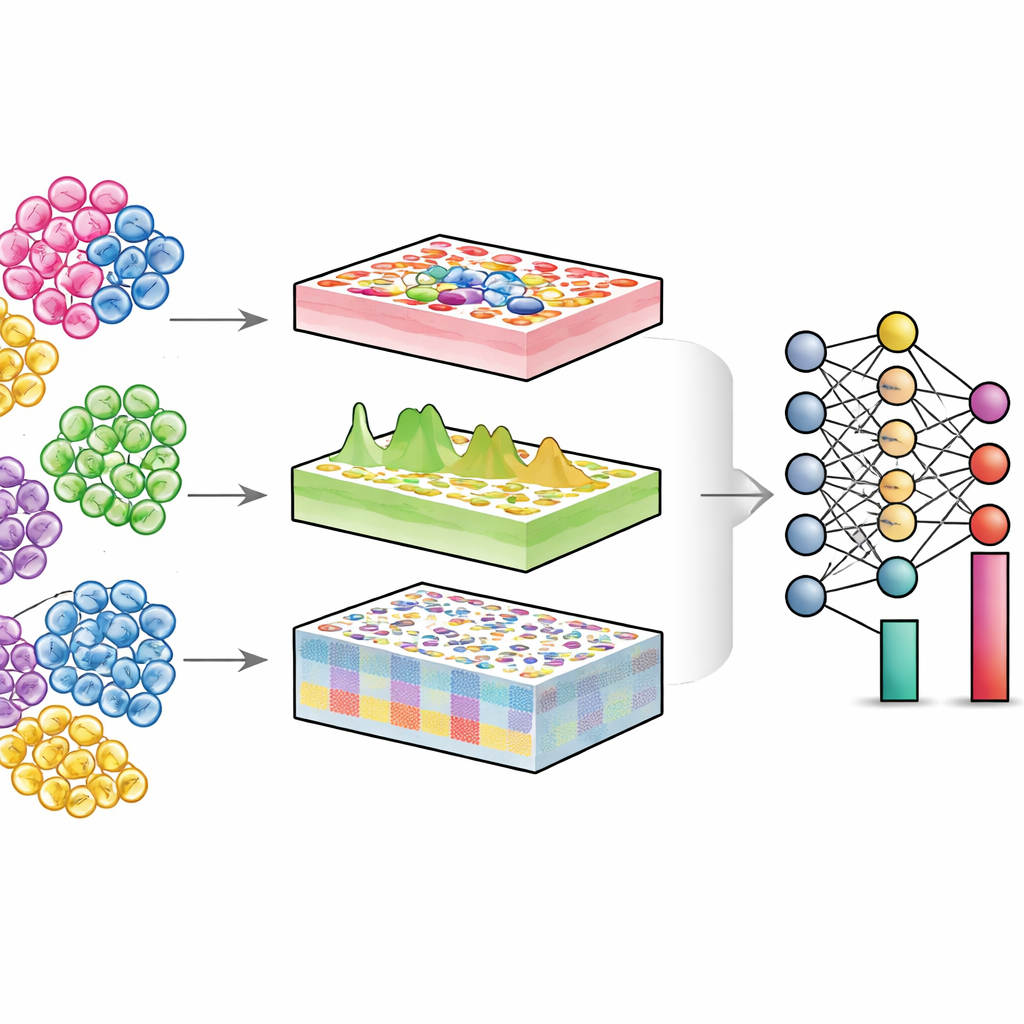
Uno sguardo all’interno dei tessuti misti
Ogni organo è una comunità di diversi tipi cellulari—cellule immunitarie, cellule strutturali, cellule staminali e altro. In condizioni di salute e malattia, ciò che spesso cambia non è solo la funzione di ciascuna cellula, ma anche quante cellule di ciascun tipo sono presenti e in quale stato si trovano. Le tecnologie single‑cell possono misurare le singole cellule direttamente, ma sono costose e tecnicamente impegnative, soprattutto per grandi coorti di pazienti o campioni conservati da tempo. Per contro, gli esperimenti “bulk” convenzionali mescolano migliaia o milioni di cellule e restituiscono un segnale medio. Gli algoritmi di deconvoluzione tentano di invertire questa mescolanza: dati i dati bulk e una mappa di riferimento single‑cell, stimano la proporzione di ciascun tipo cellulare nel tessuto.
I limiti degli strumenti specializzati
Gli strumenti di deconvoluzione esistenti sono per lo più progettati per un unico tipo di misura, come l’attività genica (trascrittomica) o le proteine (proteomica). Spesso assumono comportamenti statistici specifici che non valgono per altri tipi di dati, e incontrano difficoltà quando il tessuto bulk contiene tipi cellulari assenti nei dati di riferimento. Forti effetti batch—differenze tra donatori, strumenti o stati di salute—possono ulteriormente offuscare i segnali biologici. In particolare, non esisteva un metodo pratico per la metabolomica, lo studio delle piccole molecole che spesso sono più vicine ai sintomi clinici. Di conseguenza, gli scienziati che analizzavano coorti multiomiche dovevano usare diversi strumenti specializzati, ciascuno con le proprie peculiarità, rendendo difficile confrontare i risultati tra studi e tipi di dati.
Un motore universale di separazione
DECODE affronta queste sfide trattando la deconvoluzione come un problema flessibile di deep‑learning in grado di gestire geni, proteine e metaboliti in modo unificato. Innanzitutto sintetizza “pseudo‑tessuti” mescolando digitalmente profili single‑cell in proporzioni casuali, creando un ricco set di addestramento in cui la composizione cellulare vera è nota. Una fase di apprendimento avversario insegna quindi a un encoder a mappare sia i tessuti reali sia gli pseudo‑tessuti in una rappresentazione condivisa in cui le differenze tecniche sono minimizzate ma vengono preservati i pattern biologicamente significativi. Successivamente, un modulo speciale di denoising, guidato dall’apprendimento contrastivo, impara a separare i segnali tissutali veri dal rumore artificiale. Questo passaggio rende DECODE robusto rispetto a tipi cellulari mancanti nei riferimenti e agli errori di misura. Infine, le feature ripulite vengono passate a un modulo di deconvoluzione che stima abbondanze assolute o relative di tipi e stati cellulari, a seconda di quanto il riferimento sia completo.
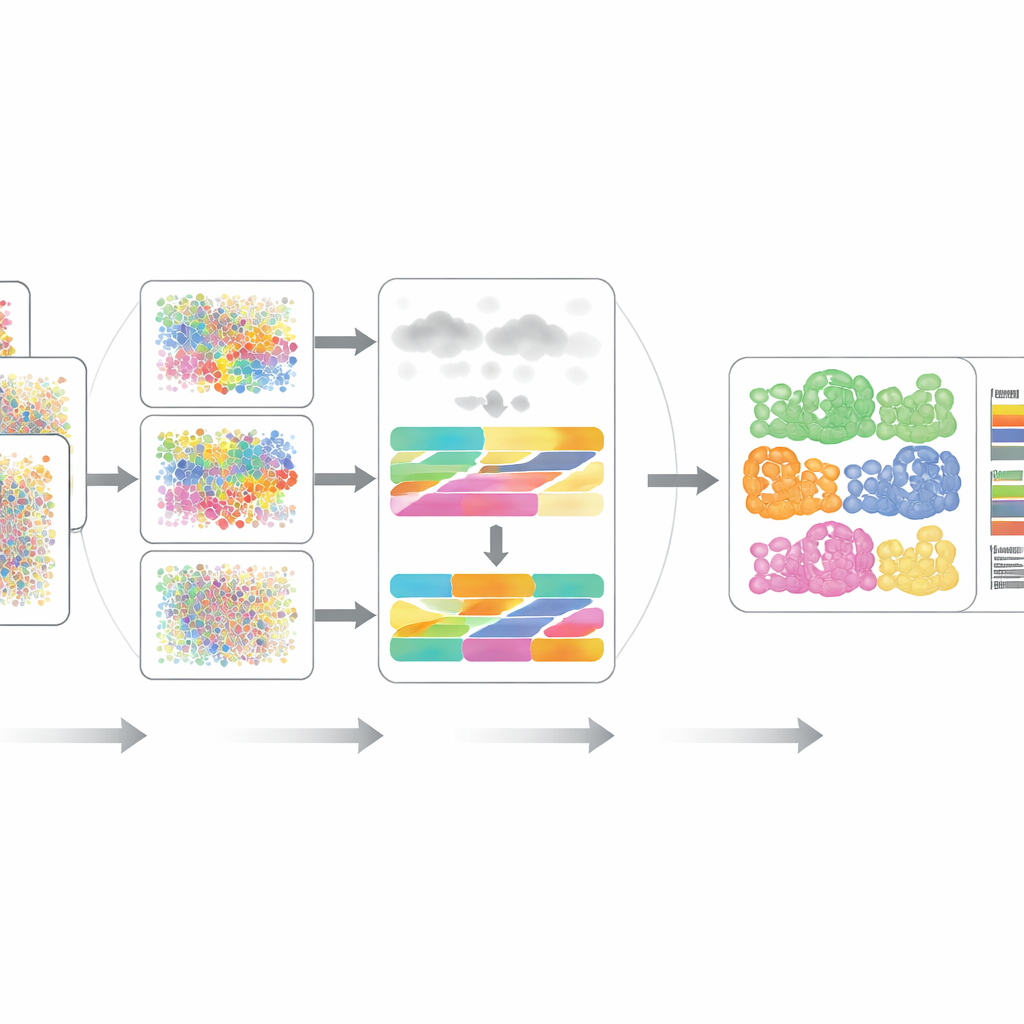
Mettere DECODE alla prova
Gli autori hanno valutato rigorosamente DECODE su 15 dataset che coprono sette scenari realistici, inclusi diversi donatori, stati di malattia, condizioni di salute, piattaforme sperimentali e persino misurazioni a risoluzione spaziale. Su trascrittomica e proteomica, DECODE generalmente ha eguagliato o superato gli strumenti allo stato dell’arte in accuratezza, mantenendo tempi di calcolo e uso della memoria ragionevoli. Crucialmente, DECODE è stato l’unico metodo a fornire risultati affidabili sui dati metabolomici, dove le feature sono meno numerose e tipi cellulari diversi possono apparire ingannevolmente simili. Il framework si è dimostrato inoltre abile nel tracciare stati cellulari—come la progressione lungo una traiettoria di sviluppo, le fasi del ciclo cellulare o le risposte a trattamenti farmacologici—invece dei soli tipi cellulari statici.
Robusto in dati del mondo reale rumorosi e incompleti
I tessuti reali spesso contengono tipi cellulari non catturati nei riferimenti single‑cell da laboratorio, e il rumore sperimentale può distorcere molte feature contemporaneamente. I ricercatori hanno simulato questi problemi aggiungendo tipi cellulari sconosciuti e introducendo diverse forme di rumore e dati mancanti in trascrittomica, proteomica e metabolomica. Nella maggior parte degli scenari, DECODE è rimasto il metodo più accurato e, in metabolomica, l’unico che non è fallito. Hanno inoltre dimostrato che DECODE fornisce risposte altamente consistenti quando applicato a misurazioni abbinate di geni e proteine dagli stessi campioni di cellule del sangue, requisito cruciale per confrontare i cambiamenti dei tipi cellulari tra layer omici in grandi coorti.
Nuove intuizioni biologiche dalle coorti multiomiche
Con questo strumento unificato, il team ha riesaminato dataset di malattie complesse. Nel cancro al seno hanno combinato coorti trascrittomiche e proteomiche per mostrare come cellule immunitarie e cellule stromali di supporto variano tra tumori non metastatici, tumori primari in metastasi e metastasi cerebrali. Pattern come una maggiore abbondanza di cellule T e di cellule perivascolari nelle lesioni non metastatiche, e l’aumento di cellule B nelle malattie avanzate, sono coerenti con studi biologici precedenti e ne estendono le conclusioni. Nel fegato murino, DECODE ha integrato coorti trascrittomiche, proteomiche e metabolomiche per seguire come epatociti, cellule endoteliali e cellule immunitarie residenti cambiano sotto diverse diete e modelli di malattia epatica, ricapitolando tendenze note come l’aumento delle frazioni di cellule di Kupffer in condizioni infiammatorie.
Cosa significa per il futuro
Per un lettore non specialista, il messaggio principale è che DECODE funziona come un prisma intelligente per i dati biomedicali: date misurazioni miscelate di tessuti, può separare i contributi di molti tipi e stati cellulari, e lo fa in modo affidabile su diversi tipi di letture molecolari. Questo consente agli scienziati di estrarre molte più informazioni dalle coorti multiomiche e dalle biobanche esistenti senza dover raccogliere nuovi dati single‑cell per ogni progetto. Pur dipendendo ancora dalla qualità e dall’ampiezza dei riferimenti single‑cell disponibili, e nonostante le risorse metabolomiche restino limitate, DECODE rappresenta un passo significativo verso l’interpretazione routinaria a livello cellulare di studi umani su larga scala, con potenziali benefici per la comprensione dei meccanismi di malattia e per la medicina di precisione.
Citazione: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Parole chiave: deconvoluzione multiomica, riferimento single-cell, deep learning in biologia, analisi metabolomica, composizione dei tipi cellulari