Clear Sky Science · it
Variazione del tasso ed errori ricorrenti nelle sequenze nella filogenetica su scala pandemica
Perché questo è importante per future epidemie
Quando un nuovo virus si diffonde in tutto il mondo, gli scienziati corrono a leggere il suo codice genetico e a ricostruirne l’albero genealogico. Quegli alberi aiutano a tracciare come emergono le varianti, quanto rapidamente si diffondono e se le misure di controllo funzionano. Ma durante il COVID-19 i laboratori hanno sequenziato milioni di genomi di SARS-CoV-2 così in fretta che errori nascosti e peculiarità nei dati hanno cominciato a distorcere il quadro. Questo articolo introduce nuovi metodi per pulire e interpretare insiemi di dati genetici così vasti, offrendo viste più chiare di come un virus pandemico evolve realmente e si muove nelle popolazioni.
La sfida di interpretare milioni di genomi
L’epidemiologia genomica trasforma i genomi virali in informazioni pratiche per le decisioni di sanità pubblica. Per SARS-CoV-2 sono stati condivisi oltre 20 milioni di genomi a livello mondiale. Gli strumenti evolutivi tradizionali sono stati sviluppati per problemi più modesti, come il confronto di geni tra specie, non per gestire milioni di sequenze virali quasi identiche che arrivano in tempo reale. Su questa scala, due problemi diventano particolarmente insidiosi. Primo, alcuni siti nel genoma virale mutano molto più frequentemente di altri, il che può far apparire simili virus non correlati. Secondo, errori tecnici ricorrenti nel sequenziamento e nell’elaborazione dei dati possono imitare mutazioni reali. Entrambi gli effetti generano “falsi echi” nell’albero evolutivo, creando incertezza su quali rami e raggruppamenti siano affidabili.
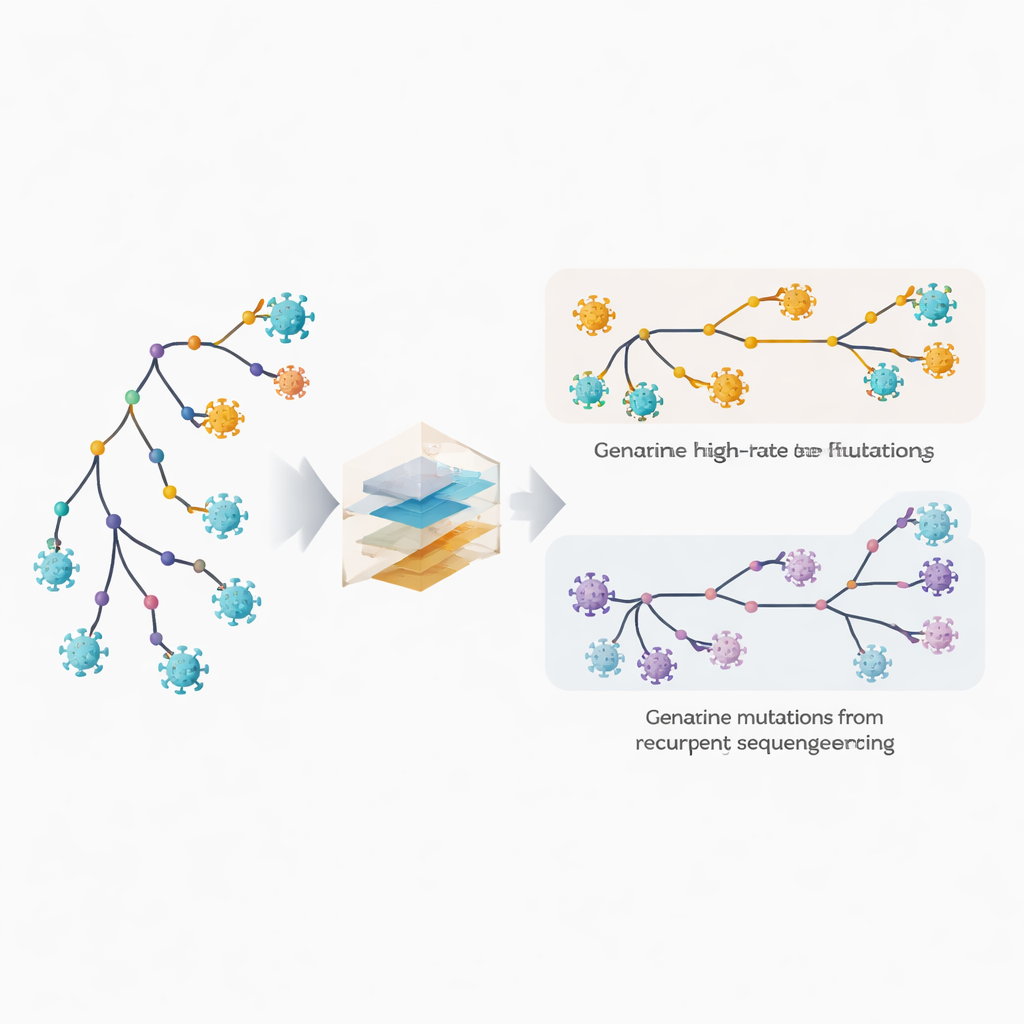
Individuare siti che cambiano rapidamente e errori nascosti
Gli autori estendono il loro software filogenetico, MAPLE, con modelli che trattano ogni posizione del genoma virale come dotata di un comportamento proprio. Invece di assumere una manciata di tassi di mutazione medi, il metodo stima un tasso separato per ogni sito, sfruttando l’enorme numero di genomi disponibili. Allo stesso tempo, permette a ogni sito di avere la propria probabilità di contenere un errore ricorrente di sequenziamento o di chiamata del consenso. Il trucco chiave è confrontare quanto spesso un cambiamento appare su rami interni profondi dell’albero, che riflettono eventi condivisi più antichi, rispetto alle punte esterne, che corrispondono a singoli genomi. Le vere mutazioni biologiche tendono a distribuirsi tra rami interni e terminali, mentre gli errori tecnici si manifestano soprattutto alle punte. Sfruttando questo schema, il metodo riesce a disaccoppiare la rapida evoluzione genuina dagli errori ripetuti.
Algoritmi più veloci per un albero della vita affollato
Gestire milioni di genomi richiederebbe normalmente una potenza di calcolo enorme. Per mantenere l’analisi pratica, il team ha riprogettato il modo in cui MAPLE memorizza e aggiorna le informazioni sulle sequenze nell’albero. Invece di confrontare ogni genoma con un singolo riferimento fisso, il software seleziona «riferimenti locali» all’interno dell’albero e registra i genomi vicini come differenze rispetto a questi punti di ancoraggio. Questa rappresentazione compatta accelera i confronti tra parti distanti dell’albero. Miglioramenti aggiuntivi affinano come nuovi campioni vengono aggiunti a un albero esistente, come vengono ottimizzate le lunghezze dei rami e come vengono esplorate forme alternative dell’albero, con opzioni per eseguire i passaggi più onerosi in parallelo su più core di processore.
Testare il metodo e ripulire i dati del mondo reale
Per verificare che i loro modelli funzionino, gli autori hanno prima creato dataset simulati realistici di SARS-CoV-2 con pattern di mutazione noti e con errori di sequenza incorporati. In questi test, il nuovo approccio ha recuperato alberi evolutivi più veritieri e individuato errori singoli con alta precisione, specialmente quando erano inclusi decine di migliaia di genomi o più. Poi si sono rivolti ai dati reali, analizzando milioni di sequenze di SARS-CoV-2 per le quali erano disponibili le letture grezze. Confrontando due diverse pipeline di costruzione del consenso, hanno identificato posizioni genomiche specifiche ripetutamente affette da artefatti, come problemi di legame dei primer o chiamate con bias verso il riferimento. Questi siti sospetti sono stati oscurati nelle analisi successive, e i genomi che mostravano segni di contaminazione o infezione mista sono stati filtrati, ottenendo un allineamento curato di oltre due milioni di sequenze di alta qualità.
Un quadro globale più chiaro dell’albero filogenetico del virus
Usando il dataset ripulito, gli autori hanno ricostruito un albero filogenetico globale di SARS-CoV-2 e mappato come le principali varianti si relazionano tra loro. Il loro albero talvolta propone relazioni leggermente diverse rispetto agli alberi pubblici precedenti, spesso in modi che richiedono meno eventi di mutazione e si adattano meglio al modello statistico. Il quadro mette anche in evidenza dove le etichette di linea possono essere incoerenti con la storia genetica sottostante, segnalando possibili ricombinanti o genomi problematici per un’ispezione più approfondita. Sebbene permangano alcune sfide — come l’overfitting quando i dati sono scarsi o l’influenza di campioni fortemente contaminati — il lavoro mostra che ora è possibile costruire alberi evolutivi su scala pandemica più affidabili. Per il lettore non specialista, la conclusione è che una migliore gestione degli errori e dei punti caldi di mutazione conduce a una comprensione più nitida di come i patogeni si diffondono e cambiano, aiutando scienziati e agenzie sanitarie a rispondere più rapidamente e con maggiore fiducia nelle future epidemie.
Citazione: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Parole chiave: genomica di SARS-CoV-2, metodi filogenetici, errori di sequenziamento, variazione del tasso di mutazione, epidemiologia genomica