Clear Sky Science · it
Atlante a singola cellula della corteccia cerebrale in sviluppo nella sindrome di Down
Perché questa ricerca è importante
La sindrome di Down è la causa genetica più comune di disabilità intellettiva, eppure sappiamo ancora poco su come alteri il cervello umano in sviluppo prima della nascita. Questo studio osserva a livello delle singole cellule la corteccia fetale — la regione cerebrale cruciale per il pensiero e la memoria — per mappare cosa va storto, quando inizia e quali interruttori molecolari potrebbero essere presi di mira per intervenire.
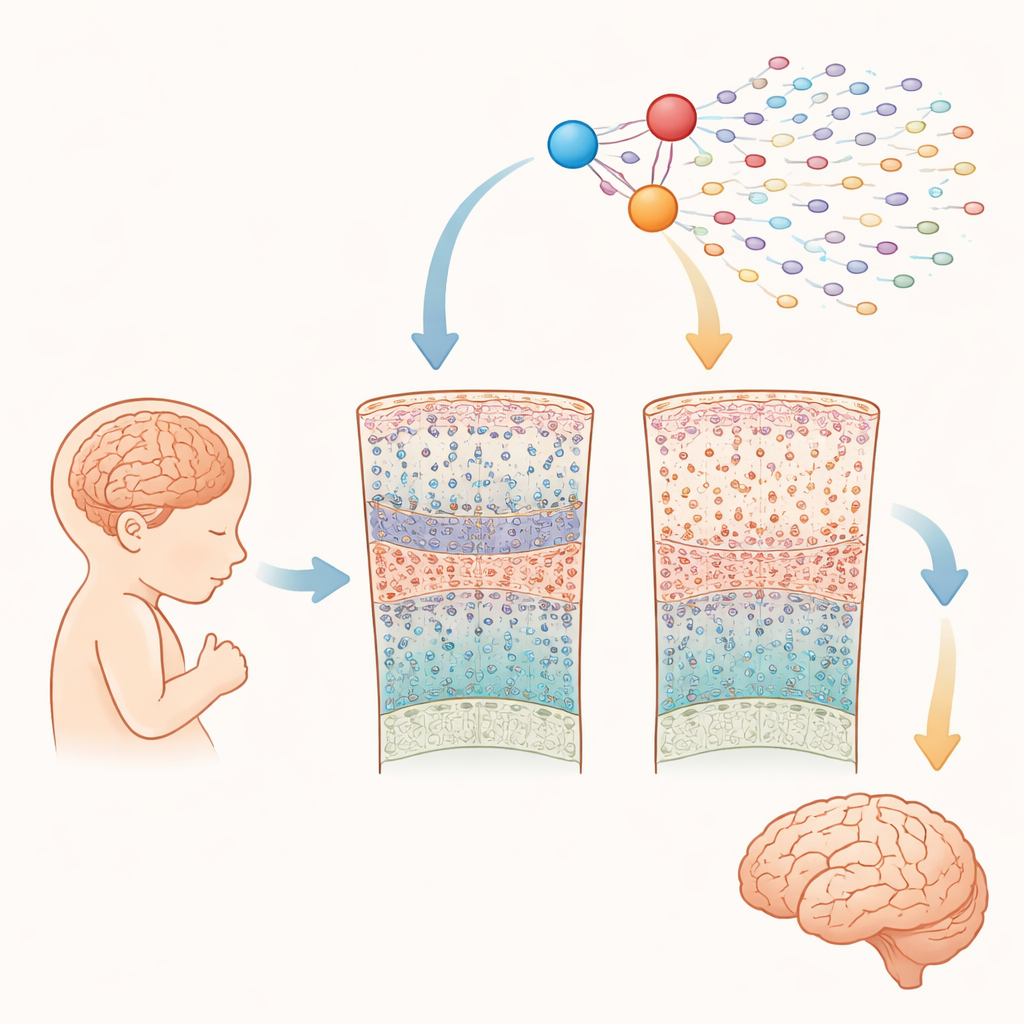
Uno sguardo ravvicinato al cervello in crescita
I ricercatori hanno analizzato quasi un quarto di milione di cellule prelevate dalla corteccia cerebrale di 15 feti con sindrome di Down e 15 senza, tra le 10 e le 20 settimane dopo il concepimento. Usando avanzate tecniche "a singola cellula", hanno misurato sia quali geni fossero attivi sia quanto fosse accessibile il DNA in ciascuna cellula. Questo ha permesso di identificare tutti i principali tipi cellulari presenti in questa fase di metà gestazione — come cellule progenitrici con caratteristiche staminali, diversi tipi di neuroni eccitatori e inibitori e le prime cellule gliali — e di confrontarne l’abbondanza e l’attività genica tra cervelli con sindrome di Down e tipici.
Cambiamenti precoci nelle cellule chiave del pensiero
La maggior parte delle classi cellulari generali era presente in numeri simili in entrambi i gruppi durante questa finestra temporale precoce. Tuttavia, il team ha riscontrato un deficit marcato e selettivo di un particolare tipo di neurone eccitatorio che normalmente occupa lo strato 4 della corteccia ed è importante per elaborare le informazioni in ingresso. Questi neuroni sono definiti dalle proteine RORB e FOXP1. Nei feti con sindrome di Down, i neuroni RORB–FOXP1 risultavano già ridotti a metà gestazione, in particolare tra le 16 e le 20 settimane, mentre altri tipi neuronali sembravano relativamente risparmiati. Ciò suggerisce che i problemi nella generazione o maturazione di questo sottogruppo di cellule iniziano in utero e possono contribuire direttamente alle difficoltà cognitive successive.
Programmi genetici interrotti e interruttori principali
Oltre ai conteggi cellulari, lo studio ha rivelato che centinaia di geni erano sottilmente dis-regolati, in particolare nei neuroni eccitatori e nei loro progenitori. Molti di questi geni sono coinvolti nella costruzione del prosencefalo, nel modellare i rami neuronali, nella formazione delle connessioni e nel supporto delle funzioni cerebrali superiori. Piuttosto che essere effetto esclusivo delle copie extra di circa 200 geni sul cromosoma 21, i dati indicano una rete di regolazione genica disturbata. Utilizzando un approccio che combina attività genica e accessibilità del DNA, gli autori hanno mappato circuiti regolatori e messo in evidenza tre fattori di trascrizione — BACH1, PKNOX1 e GABPA — codificati sul cromosoma 21 come "nodi" sensibili alla dose. Queste molecole sembrano influenzare altri regolatori critici dello sviluppo corticale, inclusi fattori già collegati alla disabilità intellettiva, e contribuiscono a spiegare come un modesto aumento di dosaggio genico di circa 1,5 volte possa riverberare attraverso interi programmi di sviluppo.
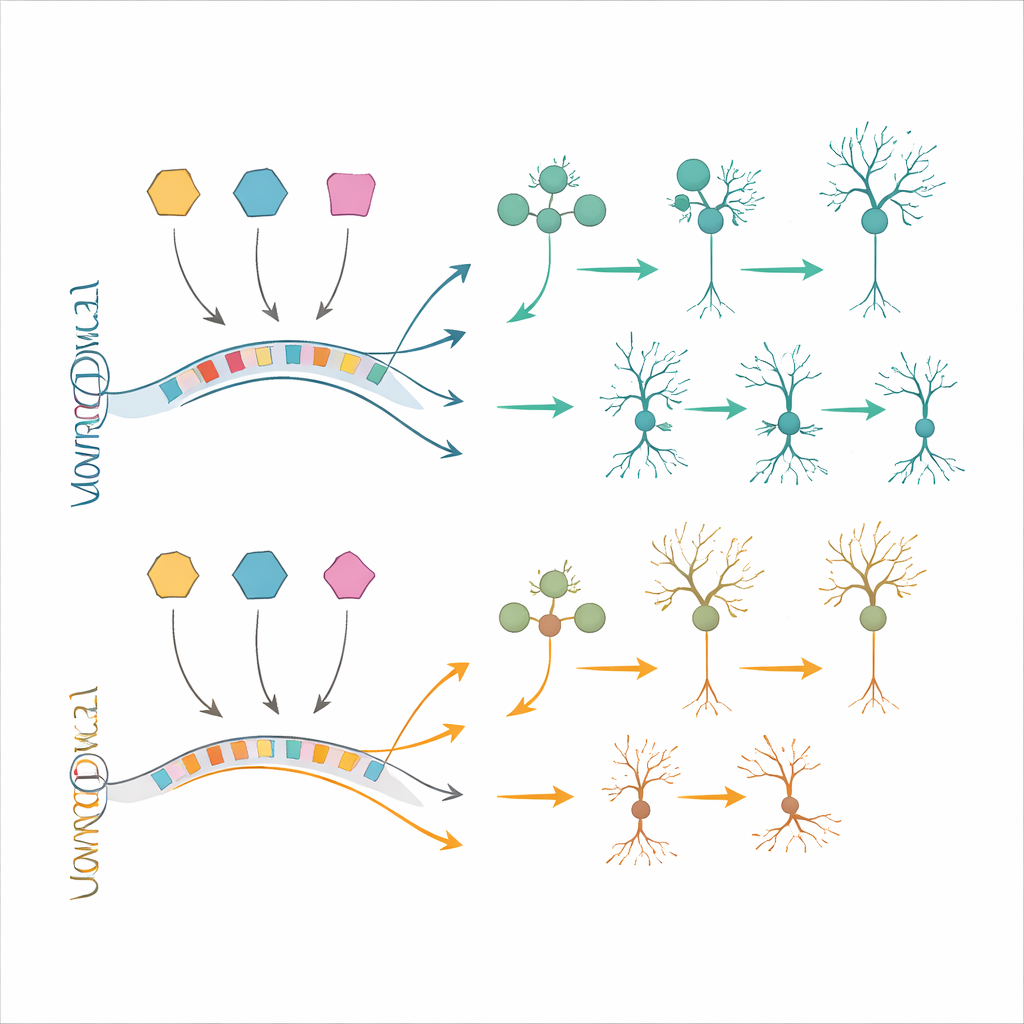
Testare strategie di recupero in cellule e cervelli viventi
Per verificare se questi cambiamenti molecolari potessero essere corretti, il gruppo ha utilizzato modelli basati su cellule staminali. Hanno generato cellule progenitrici neurali e neuroni partendo da cellule staminali pluripotenti indotte portatrici della trisomia 21 e da cellule di controllo abbinate. Molte delle modifiche nell’espressione genica osservate nei tessuti fetali sono riemerse in queste cellule coltivate in laboratorio, confermando la rilevanza dei modelli. I ricercatori hanno quindi impiegato oligonucleotidi antisenso — brevi filamenti di materiale simile al DNA ingegnerizzato — per ridurre i livelli di BACH1, PKNOX1 o GABPA verso valori normali. Questa parziale normalizzazione dei fattori di trascrizione sovraespressi ha portato a un recupero parziale di diversi geni a valle, inclusi quelli noti per essere coinvolti nella disabilità intellettiva e nella differenziazione neuronale. In un approccio complementare, hanno trapiantato cellule neurali umane con trisomia 21 in cervelli di topo e le hanno lasciate maturare in vivo. Questi innesti hanno riprodotto ulteriori caratteristiche simili alla sindrome di Down, come un equilibrio alterato tra neuroni e glia e cambiamenti genici non completamente catturati nelle colture, offrendo un banco di prova potente per future terapie.
Cosa significa per il futuro
Nel complesso, il lavoro fornisce un atlante dettagliato di come la sindrome di Down rimodelli il paesaggio genetico della corteccia in sviluppo a risoluzione a singola cellula. Per il lettore generale, il messaggio chiave è che il cromosoma extra non si limita ad aggiungere qualche gene fuori posto; modifica molti interruttori molecolari interconnessi, portando a carenze precoci e selettive di certi neuroni legati al pensiero. Individuando un piccolo insieme di fattori di trascrizione del cromosoma 21 come attori centrali — e mostrando che i loro effetti possono essere parzialmente invertiti in cellule umane — lo studio apre la strada a strategie più mirate per migliorare lo sviluppo e la funzione cerebrale nella sindrome di Down.
Citazione: Lattke, M., Tan, W.L., Sukumaran, S.K. et al. Single-cell atlas of the developing Down syndrome brain cortex. Nat Med 32, 1061–1072 (2026). https://doi.org/10.1038/s41591-026-04211-1
Parole chiave: Sindrome di Down, sviluppo cerebrale fetale, genomica a singola cellula, neuroni corticali, fattori di trascrizione