Clear Sky Science · it
Modelli trasferibili di enantioselettività a partire da dati scarsi
Un modo più intelligente per trovare il catalizzatore giusto
Spesso i chimici cercano migliori farmaci e materiali provando a unire atomi di carbonio in arrangiamenti tridimensionali molto specifici. Ottenere quel sottile risultato «destro» rispetto a «sinistro»—noto come enantioselettività—di solito significa testare molti catalizzatori metallici e condizioni di reazione per tentativi. Questo articolo introduce un metodo che usa quantità relativamente piccole di dati sperimentali, combinate con calcoli al computer rapidi, per prevedere quali catalizzatori a base di nichel daranno la mano desiderata in un’ampia gamma di reazioni, risparmiando ai chimici settimane o mesi di lavoro in laboratorio.
Perché le molecole chirali sono così difficili da controllare
Molti farmaci e prodotti naturali esistono in forme speculari che possono comportarsi in modo molto diverso nell’organismo. I catalizzatori che favoriscono un’immagine speculare rispetto all’altra sono quindi estremamente preziosi. Ma progettare tali catalizzatori è complicato. La chimica quantistica tradizionale può, in linea di principio, calcolare quale percorso preferisce una reazione, tuttavia piccoli errori energetici si traducono in grandi errori nelle previsioni di selettività, e i calcoli sono lenti. I modelli statistici più semplici, invece, sono veloci ma spesso ignorano il dettaglio dell’interazione tra il catalizzatore metallico e le molecole reagenti, specialmente quando il meccanismo di reazione può cambiare sottilmente al variare dei partner coinvolti.
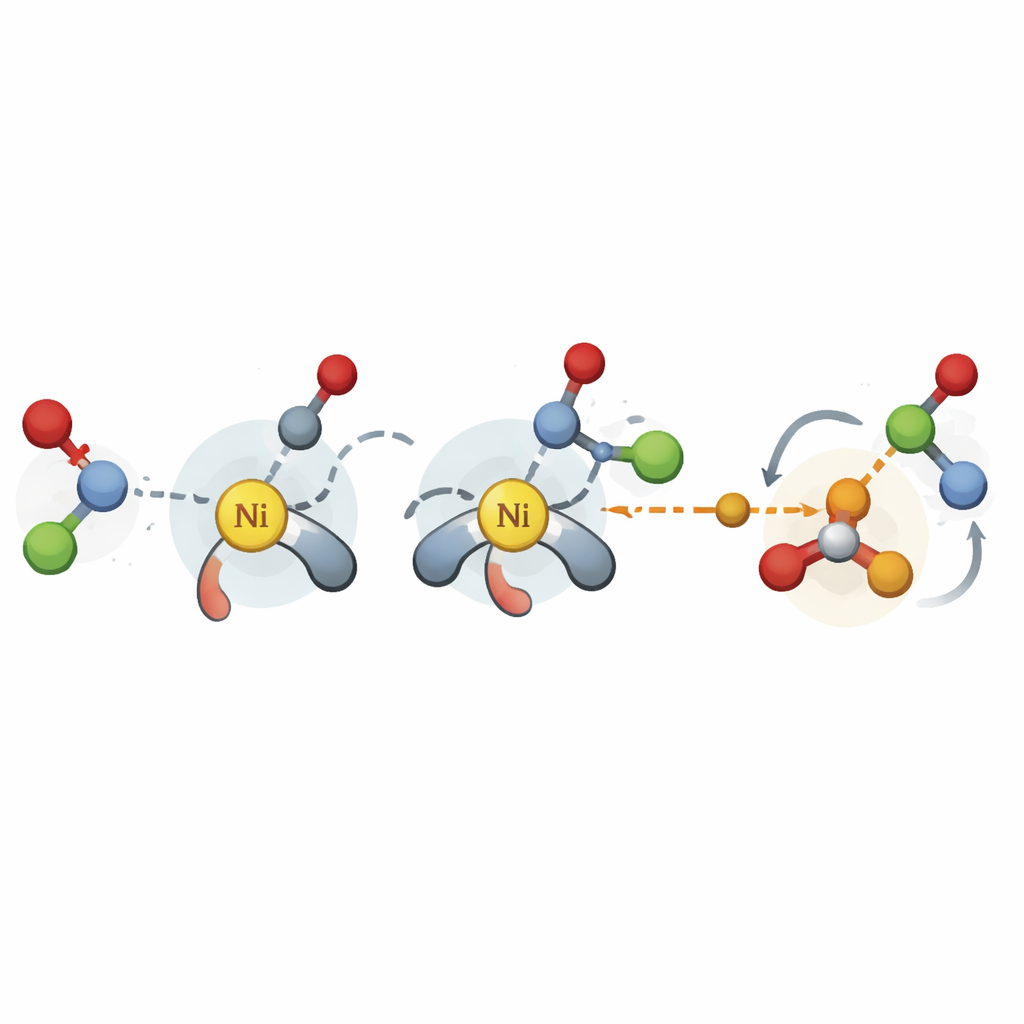
Catturare i momenti critici di una reazione
Gli autori colmano questo divario concentrandosi sulle fasi più critiche di una reazione di accoppiamento catalizzata dal nichel: i passaggi in cui si formano i nuovi legami carbonio–carbonio e in cui il prodotto finale viene rilasciato. Invece di eseguire costose simulazioni di alto livello, impiegano un metodo quantistico semplificato per generare strutture tridimensionali degli stati di transizione e degli intermedi chiave per molte possibili combinazioni di catalizzatori e substrati. Da queste strutture estraggono centinaia di descrittori fisicamente significativi, come quanto è affollato l’ambiente attorno a certi atomi o quanto facilmente possono muoversi gli elettroni. Questi numeri vengono poi inseriti in modelli di regressione lineare semplici che collegano le caratteristiche strutturali alla selettività misurata.
Imparare da dati scarsi per guidare nuovi esperimenti
Un risultato centrale del lavoro è che esso sfrutta al massimo dati scarsi—le combinazioni limitate di catalizzatori e substrati tipicamente riportate in un articolo di ricerca. In uno studio di caso, il team riesamina una reazione con nichel che accoppia ossidi di stirene con ioduri arilici. Dimostrano che i descrittori ricavati dallo stato di transizione più rilevante superano quelli ottenuti da frammenti di catalizzatore semplificati, pur essendo i calcoli sottostanti meno costosi. Con questi modelli possono testare virtualmente molti più ligandi su coppie di substrati esistenti e identificare nuove scelte di catalizzatore che aumentano l’eccesso enantiomerico per esempi particolarmente ostinati, evitando decine di esperimenti superflui.
Trasferire conoscenza tra reazioni diverse
L’approccio è potente perché può essere trasferito tra reazioni al nichel diverse ma correlate. In un secondo set di studi, gli autori combinano dati di diversi tipi di reazioni al nichel che formano tutti legami tra atomi di carbonio ibridati sp3 e partner come gruppi arilici o alchenilici, anche quando le condizioni esatte o i partner di accoppiamento differiscono. Costruendo modelli a partire dagli stessi descrittori meccanisticamente significativi, predicono con successo l’enantioselettività per nuovi ligandi, nuove combinazioni di substrati e persino per una classe interamente nuova di reazioni di formazione di legami carbonio–carbonio che non era inclusa nel set di addestramento. L’analisi dei descrittori più influenti suggerisce inoltre quale passaggio del ciclo catalitico stabilisca effettivamente la mano per ciascuna famiglia di reazioni.
Aiutare i chimici a iniziare nuove reazioni più rapidamente
In una dimostrazione finale, gli autori utilizzano il loro schema di descrittori insieme a una piattaforma di ottimizzazione bayesiana per progettare un accoppiamento catalizzato da nichel tra acetali benzigici e ioduri arilici che non era mai stato sviluppato asimmetricamente prima. Partendo dai dati in letteratura su altre reazioni, il modello suggerisce piccoli lotti di ligandi promettenti da testare, concentrandosi rapidamente sulla classe più performante in poche dozzine di esperimenti. Per un chimico, questo significa uno strumento pratico per «avviare a freddo» un nuovo progetto catalitico: inserendo una manciata di risultati iniziali, il modello può indicare quali ligandi chirali hanno più probabilità di offrire alta enantioselettività. Nel complesso, lo studio dimostra che caratteristiche computazionali selezionate con attenzione e a basso costo possono trasformare dati storici limitati in indicazioni ampiamente utili per costruire la prossima generazione di reazioni selettive.
Citazione: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Parole chiave: catalisi asimmetrica, accoppiamento incrociato con nichel, apprendimento automatico in chimica, ottimizzazione di reazione, predizione dell’enantioselettività