Clear Sky Science · it
Varianti in MTNAP1 sono all’origine di un disturbo neurodegenerativo alterando la stabilità mitocondriale
Perché questa storia è importante per la salute del cervello
Molte famiglie affrontano il dolore di vedere un bambino perdere gradualmente abilità dello sviluppo senza una diagnosi chiara. Questo studio individua una nuova causa genetica di una condizione di questo tipo, ricostruendo la catena di eventi da un singolo gene difettoso fino alle “centrali energetiche” danneggiate all’interno delle cellule cerebrali e, in ultima analisi, alla riduzione del tessuto cerebrale. Comprendere questa sequenza non solo dà risposte alle famiglie coinvolte, ma rende anche più nitida la nostra visione di quanto siano fragili i sistemi energetici del cervello.
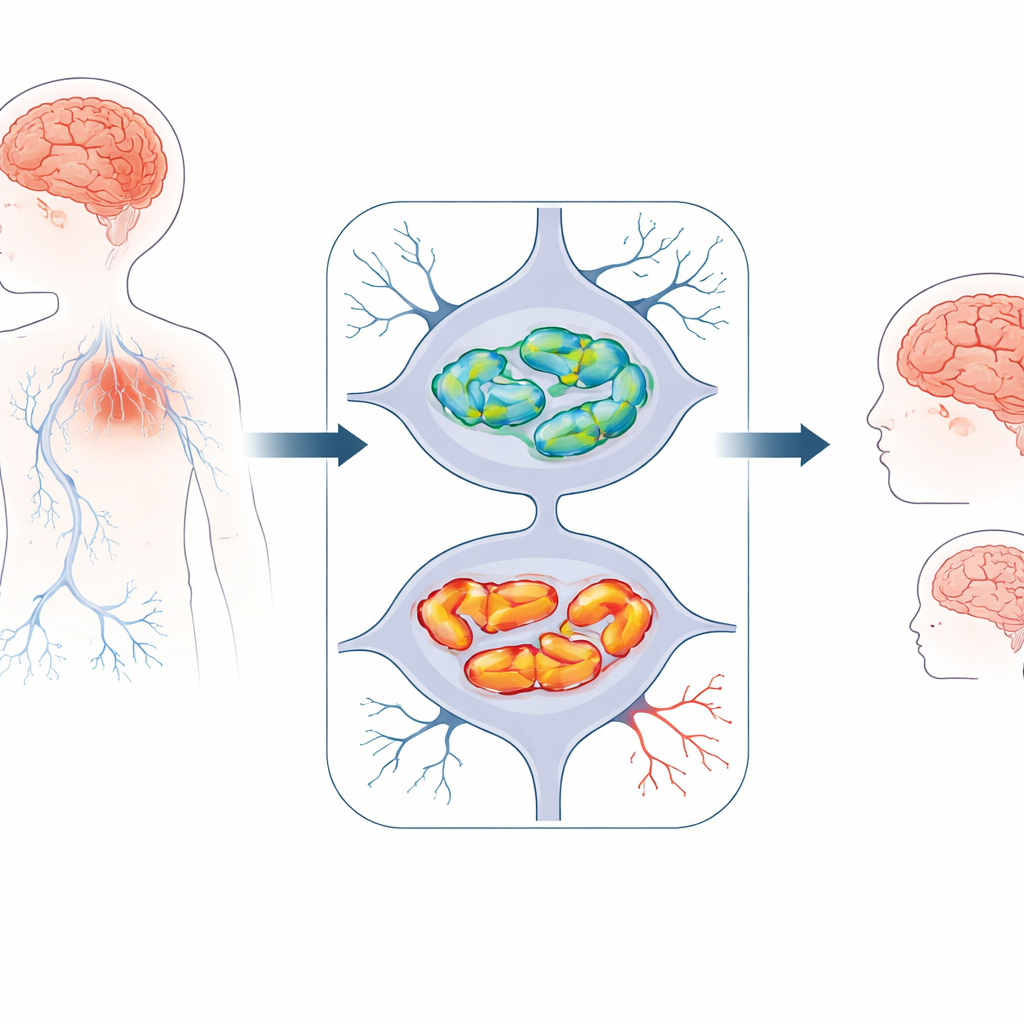
Un disturbo cerebrale infantile appena riconosciuto
I ricercatori hanno studiato tre bambini di due famiglie non correlate che presentavano problemi di sviluppo precoci. Erano più piccoli rispetto ai coetanei, hanno imparato a sedersi, camminare e parlare più tardi del previsto e poi hanno lentamente perso alcune di queste capacità. Tutti hanno sviluppato difficoltà motorie come andatura instabile, rigidità muscolare o ipotonia, e crisi epilettiche. Le immagini cerebrali hanno mostrato uno schema coerente: il tessuto sia del cervello grande (cervello) sia del “piccolo cervello” posteriore (cervelletto) si assottigliava nel tempo, e un importante ponte di fibre nervose, il corpo calloso, era insolitamente sottile. Queste caratteristiche indicavano una perdita progressiva di neuroni piuttosto che un danno singolo sull’evento della nascita.
Un gene minuscolo con grandi conseguenze
Per cercare una causa ereditaria, il gruppo ha sequenziato tutti i geni che codificano proteine nei bambini affetti e nei loro genitori. Si sono concentrati su un gene chiamato MTNAP1, che aiuta a organizzare il DNA all’interno dei mitocondri, le centrali energetiche della cellula. Ogni bambino portava due copie difettose di MTNAP1, una ereditata da ciascun genitore portatore sano. In due fratelli, un singolo cambiamento di “lettera” nel gene ha sostituito un amminoacido con un altro, alterando sottilmente la forma della proteina. Nel terzo bambino, un segnale di stop precoce nel gene probabilmente ha impedito la produzione della proteina. Queste varianti non erano presenti nelle grandi banche dati di popolazione, rafforzando l’ipotesi che si tratti di varianti rare e dannose piuttosto che di semplici varianti innocue.
Centrali energetiche sotto stress
Successivamente, gli scienziati hanno esaminato cellule della pelle prelevate dai bambini e le hanno confrontate con cellule di individui sani. Al microscopio, le cellule normali mostravano mitocondri lunghi e filamentosi che formavano una rete connessa, mentre le cellule dei bambini contenevano mitocondri corti, frammentati e aggregati. Quando i ricercatori hanno ridotto sperimentalmente i livelli di MTNAP1 in una linea cellulare di tipo neuronale umano, hanno osservato la stessa disgregazione della rete mitocondriale, confermando che la perdita di questa proteina da sola può disturbare la loro struttura. Le misurazioni dell’attività mitocondriale hanno rivelato che passaggi chiave nella produzione di energia erano compromessi e le cellule producevano un eccesso di specie reattive dell’ossigeno—sottoprodotti ossidativi dannosi che agiscono come ruggine molecolare. Le cellule sotto stress hanno smesso di dividersi correttamente, si sono accumunate in una fase di quiescenza e hanno attivato marcatori di invecchiamento precoce.
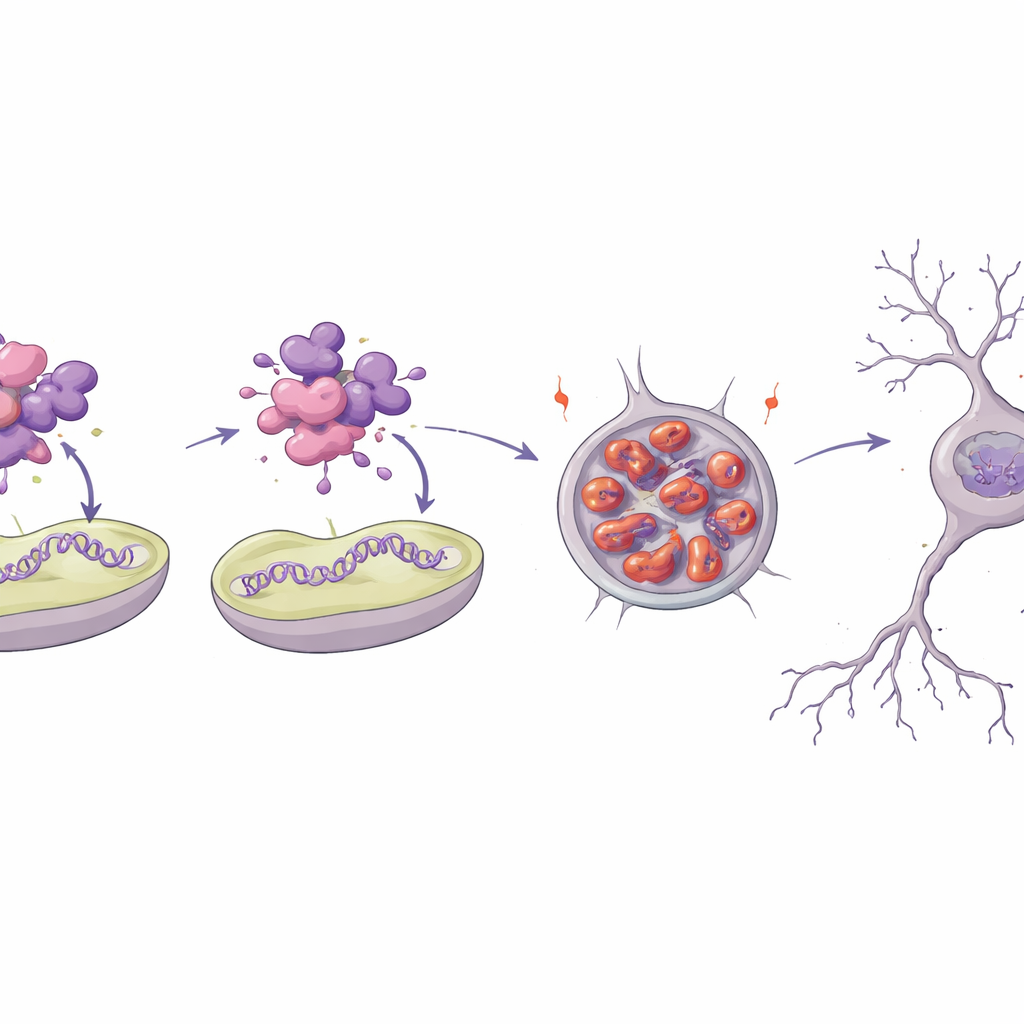
Come una singola modifica disfa una proteina vitale
Per capire perché una delle varianti sia così dannosa, il team ha modellato la struttura tridimensionale della proteina MTNAP1 e l’ha ricreata in laboratorio. L’amminoacido sostituito si trova in una regione elicoidale molto compatta che normalmente aiuta la proteina a interagire con il DNA mitocondriale e con la membrana interna. Simulazioni al computer e test biofisici hanno mostrato che la proteina mutante è meno stabile, perde gran parte della sua struttura ordinata e tende ad aggregarsi. In esperimenti in provetta, la proteina normale si legava saldamente a brevi frammenti di DNA mitocondriale e a superfici di membrana artificiali, mentre la mutante interagiva appena e invece si assemblava in aggregati di tipo amiloide. Quando introdotta in cellule di tipo neuronale, la mutante si accumulava nel tempo in grandi ammassi perinucleari, segno che i sistemi di controllo della qualità delle proteine erano sopraffatti.
Dai mitocondri danneggiati a un cervello che cede
Mettere insieme i pezzi conduce a un modello passo-passo: MTNAP1 difettoso indebolisce l’impalcatura che aiuta a organizzare il DNA mitocondriale e ad ancorare questo al compartimento membranoso interno; ciò destabilizza i mitocondri, facendoli frammentare e perdere la capacità di generare energia in modo efficiente; l’aumento dello stress ossidativo e i segnali di “invecchiamento precoce” rendono quindi i neuroni particolarmente vulnerabili, perché hanno richieste energetiche elevate e costanti e una limitata capacità di rinnovarsi. Nel cervello in sviluppo, questa crisi energetica lenta e continua si traduce in tappe di sviluppo arretrate, perdita di abilità acquisite e progressivo assottigliamento di regioni cerebrali chiave. Sebbene siano necessari altri pazienti e studi animali per mappare completamente la sindrome, questo lavoro colloca saldamente MTNAP1 come guardiano cruciale della stabilità mitocondriale e mette in evidenza l’organizzazione del DNA mitocondriale come pilastro fondamentale per uno sviluppo cerebrale sano.
Citazione: Kumar, A., Saha, S., Nasir, N. et al. Variants in MTNAP1 underlie a neurodegenerative disorder by impairing mitochondrial stability. npj Genom. Med. 11, 19 (2026). https://doi.org/10.1038/s41525-026-00554-3
Parole chiave: mitocondri, neurodegenerazione, genetica pediatrica, DNA mitocondriale, malripiegamento proteico