Clear Sky Science · it
Potenziali di apprendimento attivo per diagrammi di fase da primi principi usando nested sampling con replica-exchange
Perché questo conta per i materiali del futuro
Dai chip per computer più veloci ai componenti aeronautici più resistenti, molte tecnologie moderne dipendono dalla conoscenza di come un materiale cambia quando viene riscaldato o compresso. Questi cambiamenti, chiamati transizioni di fase, sono riassunti nei diagrammi di fase—le mappe che indicano agli scienziati quale forma di un materiale è stabile in quali condizioni. Questo studio introduce un nuovo modo per tracciare automaticamente tali mappe direttamente da calcoli quantomeccanici, utilizzando l’intelligenza artificiale per ridurre drasticamente i costi mantenendo alta accuratezza.
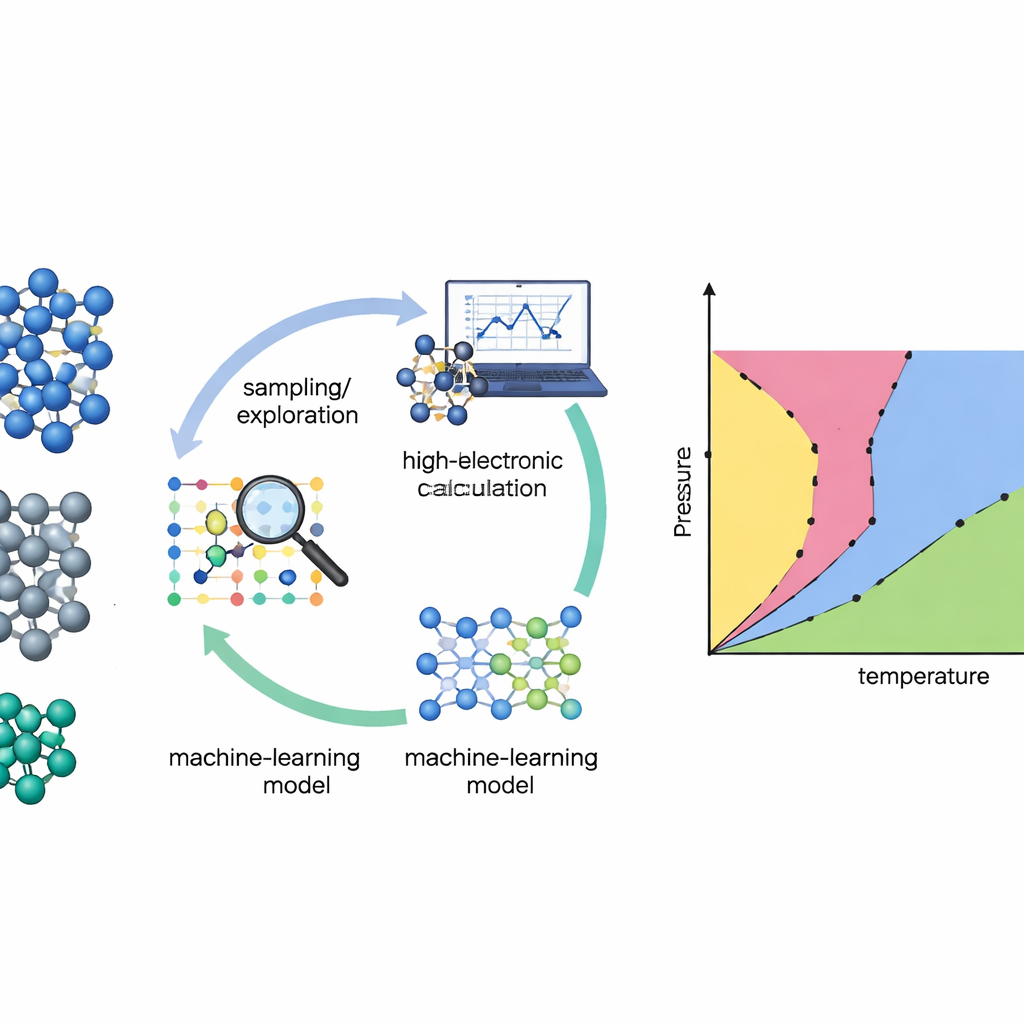
Mappare i materiali senza supposizioni
Tradizionalmente, costruire un diagramma di fase dai primi principi è come attraversare un paesaggio accidentato al buio: è necessario sospettare in anticipo dove si trovano le valli e i passi montani importanti. Molti metodi standard funzionano solo se i ricercatori forniscono forti conoscenze a priori su quali strutture cristalline o “percorsi” esplorare. Gli autori si affidano invece a una tecnica chiamata nested sampling, che setaccia in modo sistematico l’intero paesaggio energetico di un materiale senza assumere quali fasi appariranno. Seguendo quanto siano accessibili le diverse regioni di quel paesaggio, il nested sampling può ricavare proprietà termodinamiche e cambiamenti di fase su un’ampia gamma di temperature in una sola scansione.
Lasciare che il modello scelga cosa deve imparare
Anche il metodo di ricerca più intelligente ha bisogno di una buona descrizione di come gli atomi interagiscono. I calcoli quantomeccanici diretti (teoria del funzionale della densità) sono accurati ma troppo costosi per essere valutati milioni o miliardi di volte. Il gruppo affronta questo problema addestrando potenziali interatomici di machine learning—modelli rapidi che imitano le forze quantistiche tra gli atomi. Il limite è che tali modelli sono affidabili solo dove hanno visto esempi sufficienti. Per risolvere questo, gli autori costruiscono un ciclo di apprendimento attivo: il modello di machine learning esegue la simulazione di nested sampling, segnala le configurazioni in cui è incerto, e poi richiede calcoli quantomeccanici di alto livello solo su questo sottoinsieme selezionato con cura. I nuovi dati vengono reintegrati nel modello, che diventa più affidabile nelle regioni che contano di più per il diagramma di fase.
Un nuovo motore per esplorare silicio, germanio e titanio
I ricercatori hanno testato il loro approccio su tre elementi importanti: silicio e germanio, semiconduttori ben noti, e titanio, un metallo strutturale ampiamente usato. Sono partiti da banche dati iniziali modeste costruite da strutture cristalline note e semplici distorsioni, omettendo deliberatamente i liquidi e molte configurazioni ad alta energia. Il replica-exchange nested sampling—molte esecuzioni di nested sampling a diverse pressioni che possono scambiarsi configurazioni—ha quindi esplorato i paesaggi energetici dei materiali. Dopo ogni ciclo di esplorazione, l’algoritmo ha selezionato automaticamente centinaia di configurazioni atomiche rappresentative, pesando verso quelle in cui le previsioni delle forze differivano maggiormente all’interno di un comitato di modelli a rete neurale. Queste sono state ricalcolate con un metodo quantomeccanico ad alta accuratezza (r2SCAN) e usate per ritrainare i potenziali prima di avviare il ciclo successivo.
Dai primi rumori a mappe di fase affidabili
In circa dieci-quindici cicli di apprendimento, l’incertezza dei modelli si è progressivamente ridotta, specialmente nelle forze che governano il moto atomico. Allo stesso tempo, le traiettorie del nested sampling hanno iniziato a rivelare i contorni familiari dei diagrammi di fase. Per il silicio, il metodo ha riprodotto la nota struttura a diamante a bassa pressione, la sua fase esagonale ad alta pressione e il caratteristico comportamento di fusione con temperatura e pressione, tutto in buon accordo con esperimenti e simulazioni precedenti. Il germanio ha mostrato un andamento simile, con una fase tipo diamante a bassa pressione che cede il passo a una fase metallica ad alta pressione, sebbene la pressione di transizione esatta fosse spostata in parte a causa dell’approssimazione quantomeccanica scelta. Il titanio ha rappresentato una prova più difficile: le sue fasi sono metalliche, strutturalmente simili e separate da piccole differenze energetiche. Anche lì, la strategia di apprendimento attivo ha catturato la sequenza delle fasi solide e la linea di fusione, e controlli aggiuntivi basati sulle funzioni di distribuzione radiale hanno confermato l’identità delle strutture predette.
Cosa significa questo per progettare nuovi materiali
In termini semplici, lo studio dimostra che un computer può ormai insegnarsi come si comporta un materiale su un’ampia gamma di temperature e pressioni, interrogando un “oracolo” quantomeccanico solo quando necessario. Il motore replica-exchange nested sampling garantisce un’esplorazione ampia e non distorta, mentre il ciclo di apprendimento attivo assicura che i potenziali di machine learning siano accurati dove conta termodinamicamente. Anche se il lavoro corrente si concentra su tre elementi e un particolare metodo quantomeccanico, il quadro è generale: può essere abbinato a teorie elettroniche più avanzate o a reti neurali potenti, ed esteso ad leghe complesse o composti. Con il miglioramento della potenza di calcolo e degli algoritmi, questo tipo di flusso di lavoro autonomo potrebbe diventare uno strumento standard per prevedere diagrammi di fase e guidare la scoperta di nuovi materiali con proprietà mirate.
Citazione: Unglert, N., Ketter, M. & Madsen, G.K.H. Active learning potentials for first-principles phase diagrams using replica-exchange nested sampling. npj Comput Mater 12, 107 (2026). https://doi.org/10.1038/s41524-026-01989-z
Parole chiave: diagrammi di fase dei materiali, apprendimento attivo, potenziali di machine learning, nested sampling, silicio germanio titanio