Clear Sky Science · it
Mescolamento spaziale efficiente e accurato di potenziali interatomici appresi da macchine per la scienza dei materiali
Perché contano simulazioni atomiche più veloci
Progettare materiali migliori per tecnologie come la fusione nucleare, la microelettronica e le leghe strutturali si basa sempre più su simulazioni al computer che seguono il movimento e le interazioni degli atomi. I metodi più accurati prendono spunto dalla fisica quantistica, ma sono così costosi dal punto di vista computazionale che sono praticabili solo su dimensioni di sistema e scale temporali modeste. Questo articolo presenta ML‑MIX, una tecnica e un pacchetto software che consentono ai ricercatori di mantenere l’accuratezza vicino a quella quantistica esattamente dove serve, usando modelli più semplici ed economici altrove. Il risultato è un aumento sostanziale della velocità—spesso di un fattore 4–10—senza perdere affidabilità nelle predizioni fisiche chiave.
Fondere viste dettagliate e semplificate degli atomi
Al centro del lavoro c’è un’idea semplice: non tutti gli atomi in una simulazione richiedono lo stesso livello di attenzione. Le regioni in cui i legami si allungano, si rompono o si riorganizzano—come difetti, superfici o particelle impiantate—beneficiano di potenziali interatomici basati su apprendimento automatico, che riproducono l’accuratezza quantomeccanica. Ma gli atomi lontani da questi “punti caldi” vibrano per lo più attorno a posizioni regolari e possono essere trattati con modelli molto più semplici. ML‑MIX fornisce un modo per combinare un modello accurato ma costoso con uno più snello e “economico” all’interno della stessa scatola di simulazione. Lo fa definendo una zona core che usa il modello costoso, un buffer circostante in cui le forze vengono miscelate con cura tra i modelli, e una zona bulk esterna che utilizza solo la descrizione economica. 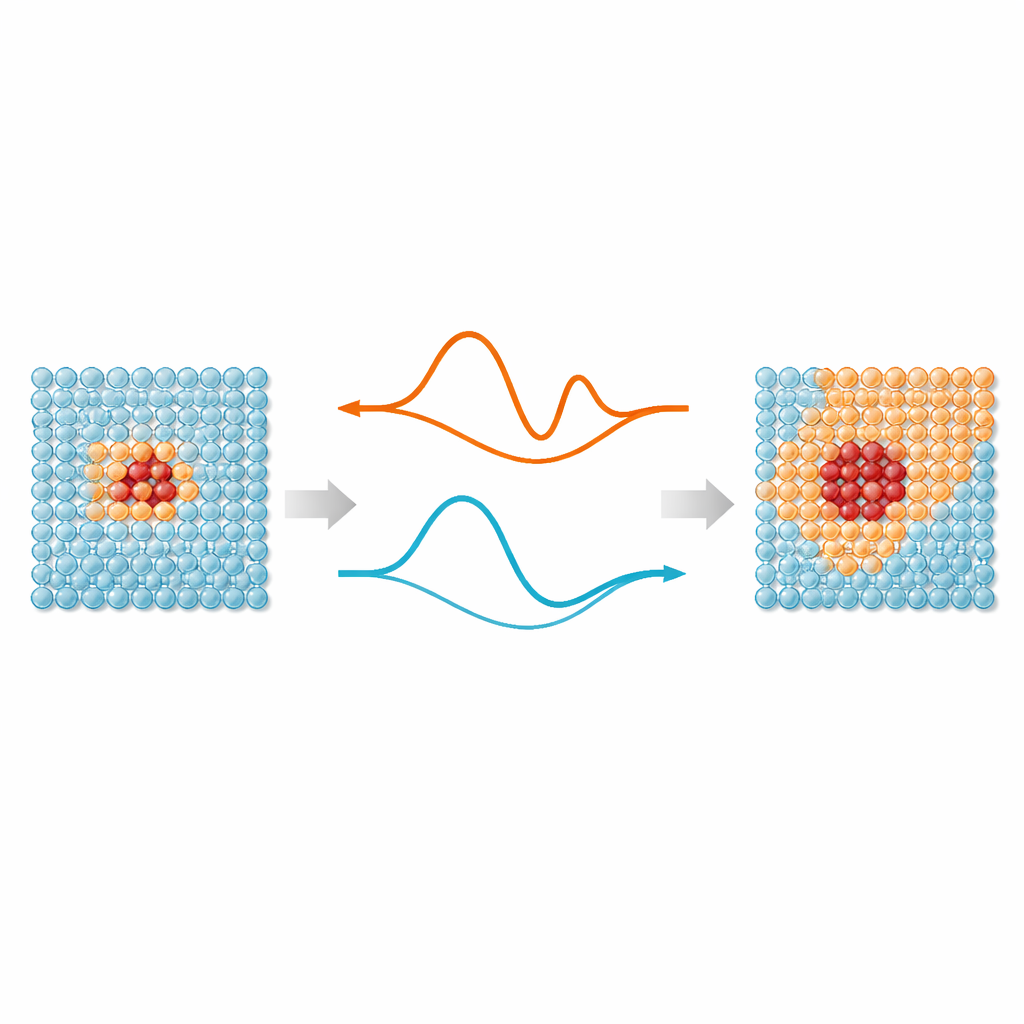
Insegnare a un modello economico a imitare uno accurato
Una sfida chiave è assicurare che il modello economico si comporti come quello accurato ovunque si incontrino. Invece di adattare il modello economico direttamente a un vasto e variegato insieme di dati quantomeccanici, gli autori generano dati “sintetici” mirati eseguendo il modello accurato nelle condizioni specifiche rilevanti per la regione bulk: vibrazioni ad alta temperatura e cristalli leggermente deformati. Poi adattano il modello economico in modo che corrisponda a questi dati, imponendo al contempo vincoli rigorosi su proprietà materiali di base come le costanti elastiche e la spaziatura reticolare. Questo fitting vincolato garantisce che le tensioni e le deformazioni a lungo raggio combacino in modo fluido attraverso il confine fra i due modelli, evitando forze artificiali che potrebbero corrompere la dinamica vicino all’interfaccia.
Mettere il metodo alla prova
Per verificare che ML‑MIX funzioni davvero, gli autori eseguono una serie di test su sistemi di silicio, ferro e tungsteno. Per un esempio semplice, calcolano la barriera energetica per lo spostamento di una vacanza—un sito reticolare vuoto—nel silicio da una posizione all’altra. La simulazione mista riproduce il risultato di una simulazione completamente con il modello costoso con una precisione di circa un millesimo di elettron‑volt, correndo però circa cinque volte più veloce. In un contesto più dinamico, allungano un singolo legame di silicio in un cristallo caldo e misurano la forza media su di esso. Una simulazione che usa solo il modello economico si avvicina già sorprendentemente, ma una volta aggiunto un piccolo core costoso attorno al legame allungato, l’accordo diventa statisticamente indistinguibile dal riferimento completamente accurato, con accelerazioni fino a circa un fattore 13 in esecuzioni seriali.
Seguire in movimento difetti e particelle
Test più realistici esaminano come si muovono i difetti nei metalli. Il gruppo simula la diffusione di un difetto auto‑interstiziale nel ferro e di atomi di elio all’interno del tungsteno. In entrambi i casi, il modello costoso è confinato a una piccola regione mobile attorno al difetto, mentre il resto del cristallo è gestito dal potenziale economico. I coefficienti di diffusione risultanti corrispondono a quelli delle simulazioni completamente accurate entro l’errore statistico, anche quando una simulazione solo‑economica fallirebbe. Gli autori poi spingono il metodo su problemi più grandi e scientificamente importanti nel tungsteno, un candidato di primo piano per i reattori a fusione. Modellano il moto di dislocazioni elicoidali—difetti lineari che controllano la deformazione plastica—e l’impianto di atomi di elio in una superficie di tungsteno calda. In entrambi i casi, ML‑MIX riproduce i risultati ottenuti solo con il modello costoso riducendo il costo computazionale di fattori dell’ordine di circa quattro‑undici. 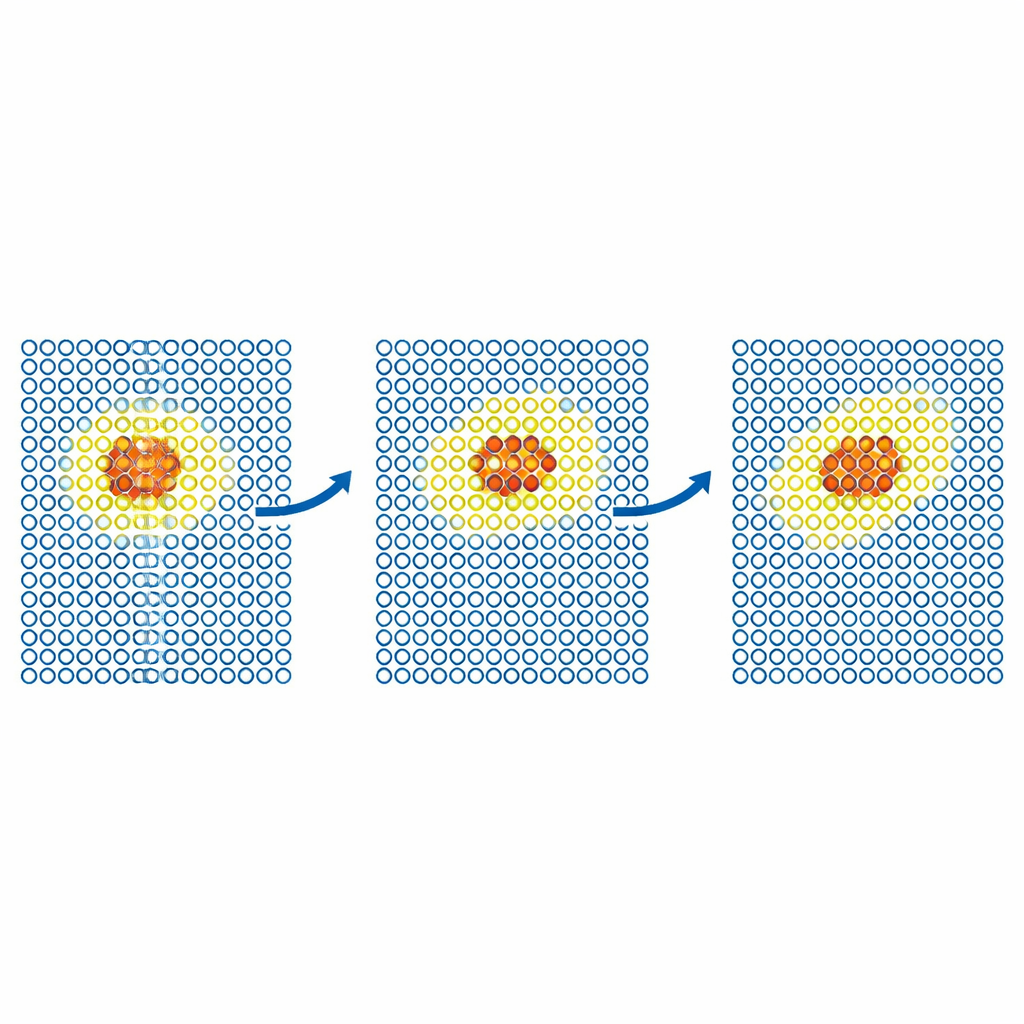
Corrispondenza con gli esperimenti e prospettive
Lo studio sull’impianto di elio mostra più chiaramente il potenziale di questo approccio. Mescolando un modello all’avanguardia basato sull’apprendimento automatico per le interazioni elio–tungsteno con un potenziale più veloce per il tungsteno puro, gli autori simulano molti più eventi d’urto e campioni più grandi di quanto sarebbe altrimenti fattibile, il tutto su processori grafici. La frazione prevista di atomi di elio che rimbalzano dalla superficie rispetto a quelli che si impiantano nel metallo è in accordo con misure sperimentali fino a energie di impatto di circa 80 elettronvolt, un risultato che simulazioni precedenti faticavano a ottenere. Sebbene lo schema di mescolamento non conservi strettamente l’energia e richieda termostati delicati, la deriva risultante è piccola e gestibile. Nel complesso, ML‑MIX dimostra che combinare con cura modelli atomici dettagliati e semplificati può superare barriere di lunga data tra accuratezza e scala, aprendo la strada a simulazioni di routine ad alta fedeltà di materiali complessi in ambienti realistici.
Citazione: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Parole chiave: potenziali interatomici appresi da macchine, simulazione multiscala dei materiali, impianto di elio nel tungsteno, difetti e dislocazioni, accelerazione della dinamica molecolare