Clear Sky Science · it
Espansione di cluster atomici a grafo per potenziali interatomici fondamentali basati su machine learning
Insegnare ai computer a percepire gli atomi
Progettare nuovi materiali per batterie, aeroplani o reattori a fusione spesso si riduce a una domanda semplice: come si spingono e si attraggono gli atomi? Calcolare queste forze esattamente è così costoso che può richiedere giorni su un supercomputer per un singolo materiale. Questo articolo presenta una nuova famiglia di modelli di machine learning, chiamata GRACE, che funziona come una “calcolatrice” universale per le forze atomiche su gran parte della tavola periodica. L’obiettivo è rendere le simulazioni accurate di materiali complessi routine anziché imprese eroiche.
Un modello unico per molti materiali
La maggior parte dei campi di forza basati su machine learning esistenti sono strumenti specialistici: funzionano molto bene per pochi elementi o composti, ma devono essere ricostruiti da zero quando si aggiungono nuovi elementi. GRACE sceglie una via diversa. È progettato fin dall’inizio come modello fondamentale in grado di gestire 89 elementi chimici e un’enorme varietà di disposizioni atomiche con un unico insieme condiviso di regole. Per farlo, gli autori si basano su un quadro matematico chiamato espansione di cluster atomici e lo estendono a strutture simili a grafi, permettendo al modello di descrivere sia i vicinati locali degli atomi sia schemi più estesi in modo unificato. Invece di codificare a mano ogni possibile interazione, GRACE apprende “incapsulamenti” compatti che catturano somiglianze tra gli elementi, in modo che la conoscenza su un materiale possa aiutare a descriverne un altro.
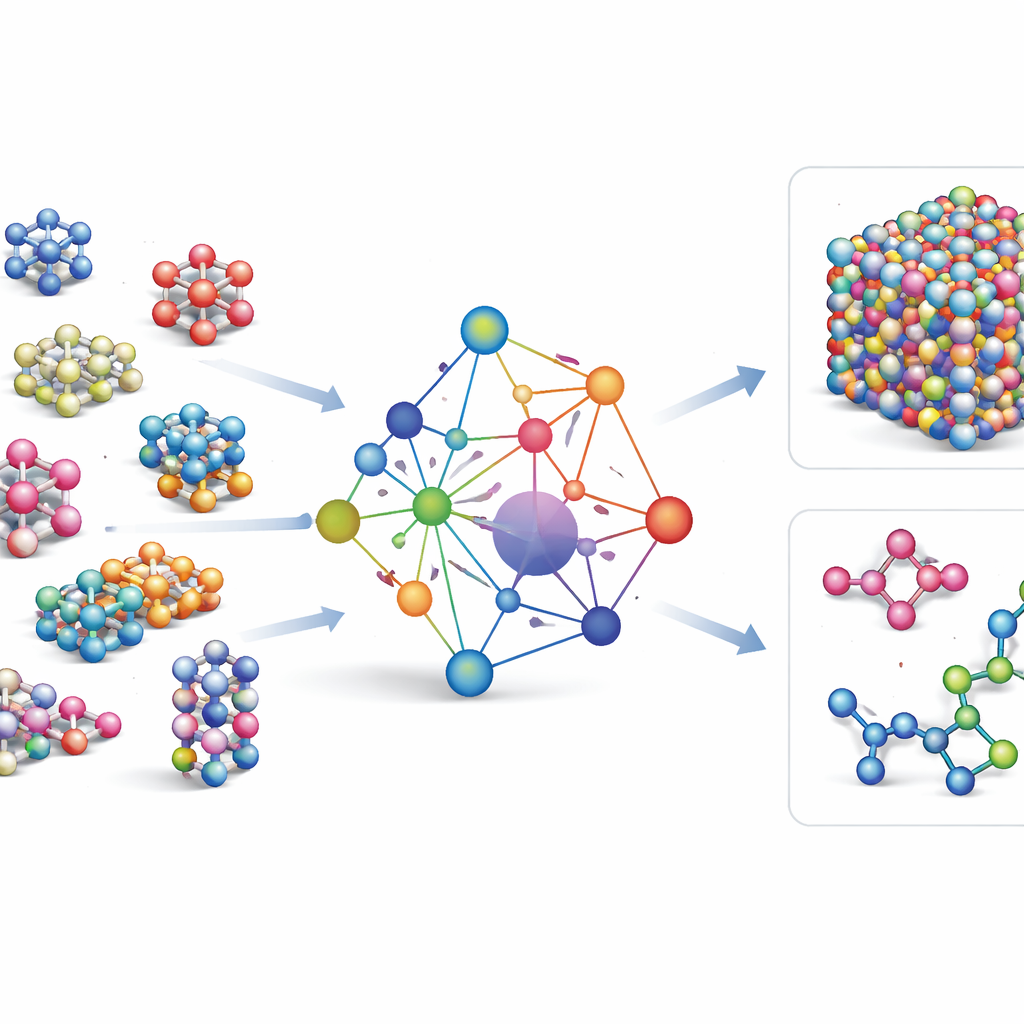
Addestrare su un mare di dati atomici
Per insegnare a GRACE come si comportano gli atomi, gli autori hanno assemblato alcuni dei più grandi database pubblici di calcoli quantomeccanici. Il nucleo è la raccolta OMat24, che contiene circa 110 milioni di simulazioni di materiali inorganici, integrata da altre due raccolte che tracciano come le strutture si rilassano ed evolvono. Nel loro insieme, questi set di dati coprono cristalli vicini all’equilibrio, strutture deformate e distorte, liquidi ad alta temperatura e altro ancora, su lo stesso ampio insieme di elementi. I modelli GRACE esistono in diverse dimensioni, da versioni più semplici a uno strato che considerano solo l’ambiente atomico locale, fino a versioni più profonde a due strati che effettivamente trasferiscono “messaggi” tra regioni vicine. L’addestramento iniziale mira a un buon equilibrio tra energie, forze e tensioni interne, e un affinamento successivo regola i modelli per renderli compatibili con database di riferimento ampiamente usati nella scienza dei materiali.
Mettere il modello alla prova
Un modello universale è utile solo se funziona in modo affidabile su molti compiti. Gli autori quindi hanno sottoposto GRACE a una batteria di test impegnativa che rispecchia come gli scienziati usano realmente le simulazioni atomistiche. In un benchmark di comunità per la scoperta di strutture cristalline stabili, GRACE si trova costantemente sul “fronte di Pareto”: per una data accuratezza, è più veloce dei modelli concorrenti, e per una data velocità è più accurato. Vantaggi simili emergono nella previsione della conducibilità termica, una proprietà molto sensibile a piccole variazioni nel moto atomico. GRACE si comporta bene anche sulle proprietà elastiche, sulle energie di superficie, sulle energie di bordo di grano e sulle energie di formazione di difetti puntiformi in molti metalli puri, tutti test che sondano come i materiali rispondono a trazione, taglio o danni locali. Una lunga simulazione di dinamica molecolare di un sale fuso caldo mostra che il modello rimane numericamente stabile per nanosecondi riproducendo al contempo dettagliati modelli strutturali e tassi di diffusione atomica.
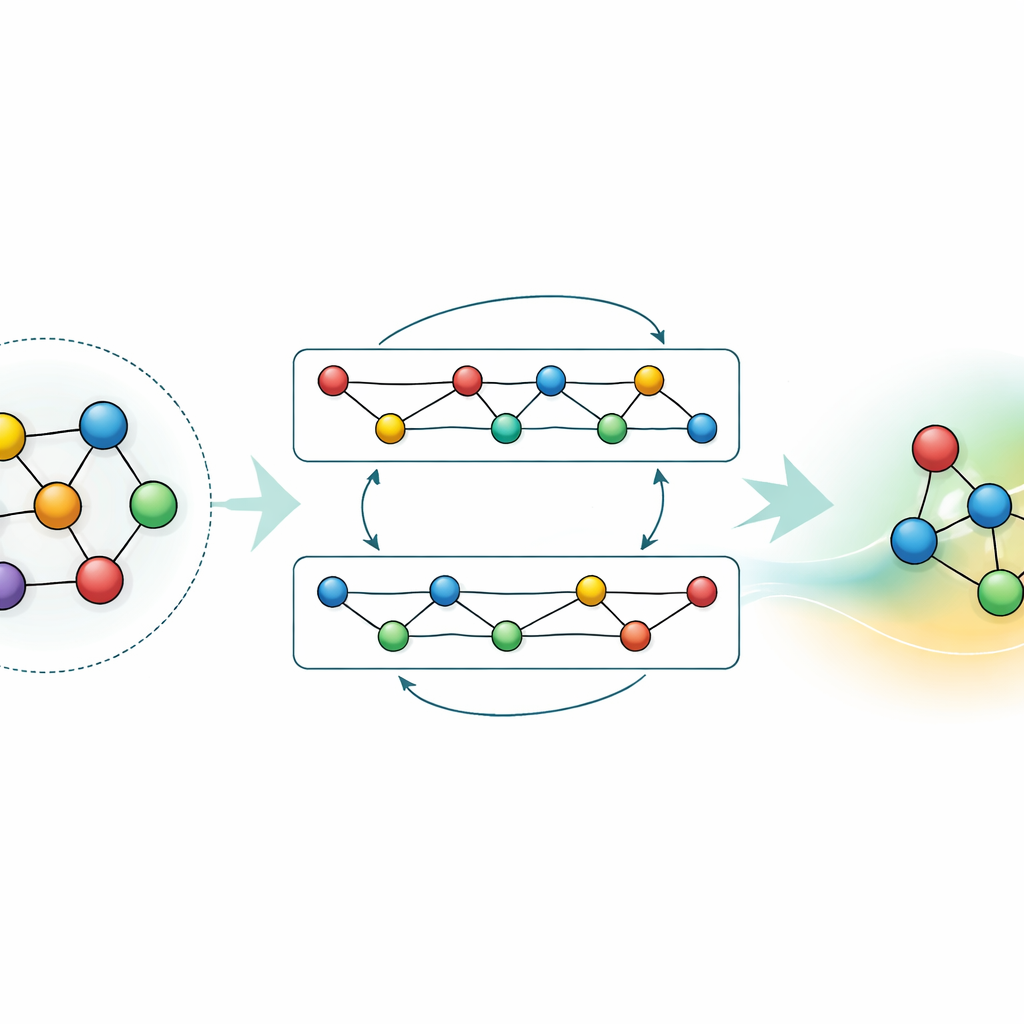
Adattare e comprimere la conoscenza
Pur essendo potente, un modello a uso generale necessita in molte applicazioni o di maggiore accuratezza per un materiale specifico o di calcoli più veloci su hardware modesto. Gli autori dimostrano due strategie per ottenere questo senza buttare via ciò che GRACE ha già imparato. Prima, affinano il modello fondamentale su dataset mirati, come leghe alluminio‑litio o percorsi dettagliati di combustione dell’idrogeno. Per le leghe, anche dati aggiuntivi modesti migliorano significativamente le previsioni, superando modelli addestrati da zero con le stesse informazioni. Per la combustione, un fine‑tuning ingenuo causerebbe normalmente che il modello “dimentichi” ciò che sapeva sugli altri materiali; congelando in modo selettivo parti della rete e aggiornando solo parametri scelti, gli autori limitano questo fenomeno di dimenticanza catastrofica pur ottenendo maggiore accuratezza per la nuova chimica. Secondo, mostrano come distillare il grande modello in un “studente” molto più semplice che imiti l’insegnante su sistemi chiave. Questa versione distillata gira circa settanta volte più veloce su CPU pur preservando gran parte dell’accuratezza, specialmente quando addestrata su un mix di leghe complesse e strutture di riferimento più semplici etichettate dall’originale GRACE.
Che cosa significa per il design futuro dei materiali
Il lavoro colloca GRACE come una base flessibile per la prossima generazione di modellazione atomistica. Invece di creare un nuovo potenziale per ogni materiale o proprietà, i ricercatori possono partire da un modello universale GRACE e poi affinarlo o destillarlo secondo le proprie esigenze, risparmiando enormi quantità di tempo di calcolo e lavoro di esperti. I benchmark mostrano che questo approccio non si limita a uguagliare gli strumenti esistenti; spesso li supera sia in velocità sia in affidabilità, in particolare per proprietà esigenti come il trasporto termico. Per i non specialisti, il messaggio chiave è che un singolo modello di machine learning ben progettato può ora fungere da “motore” ampiamente affidabile per esperimenti virtuali su gran parte della tavola periodica, accelerando la ricerca di batterie migliori, catalizzatori, leghe strutturali e materiali per l’energia.
Citazione: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Parole chiave: potenziali interatomici con machine learning, modellazione dei materiali, simulazioni atomiche, modelli fondamentali, espansione di cluster atomici a grafo