Clear Sky Science · it
Flusso di lavoro automatizzato assistito da potenziali di apprendimento automatico auto-ottimizzanti per la progettazione di materiali complessi altamente efficiente
Ricerche più intelligenti per nuovi materiali
Progettare nuovi materiali è un po’ come cercare un ago in un pagliaio quasi infinito. Dalle batterie migliori e computer più veloci a laser più efficienti e potenziali superconduttori a temperatura ambiente, molte tecnologie future dipendono dalla scoperta delle giuste disposizioni atomiche. Questo articolo presenta un modo per lasciare all’intelligenza artificiale la maggior parte di quella ricerca in modo automatico, riducendo drasticamente tempo e costi necessari per trovare composti promettenti.
Perché il rompicapo dei materiali è così difficile
Le proprietà di un solido — quanto bene conduce l’elettricità, quanto è resistente, come risponde alla luce — sono determinate da come i suoi atomi sono disposti in schemi tridimensionali chiamati strutture cristalline. In teoria, si possono usare i principi della meccanica quantistica per calcolare quali disposizioni sono stabili e quali saranno le loro proprietà. In pratica, questi calcoli quantistici sono così onerosi che solo una piccolissima frazione di tutti i materiali possibili può essere verificata. La sfida cresce rapidamente quando sono coinvolti più di due elementi chimici, perché il numero di combinazioni e di disposizioni atomiche esplode, rendendo impossibile una ricerca alla cieca.
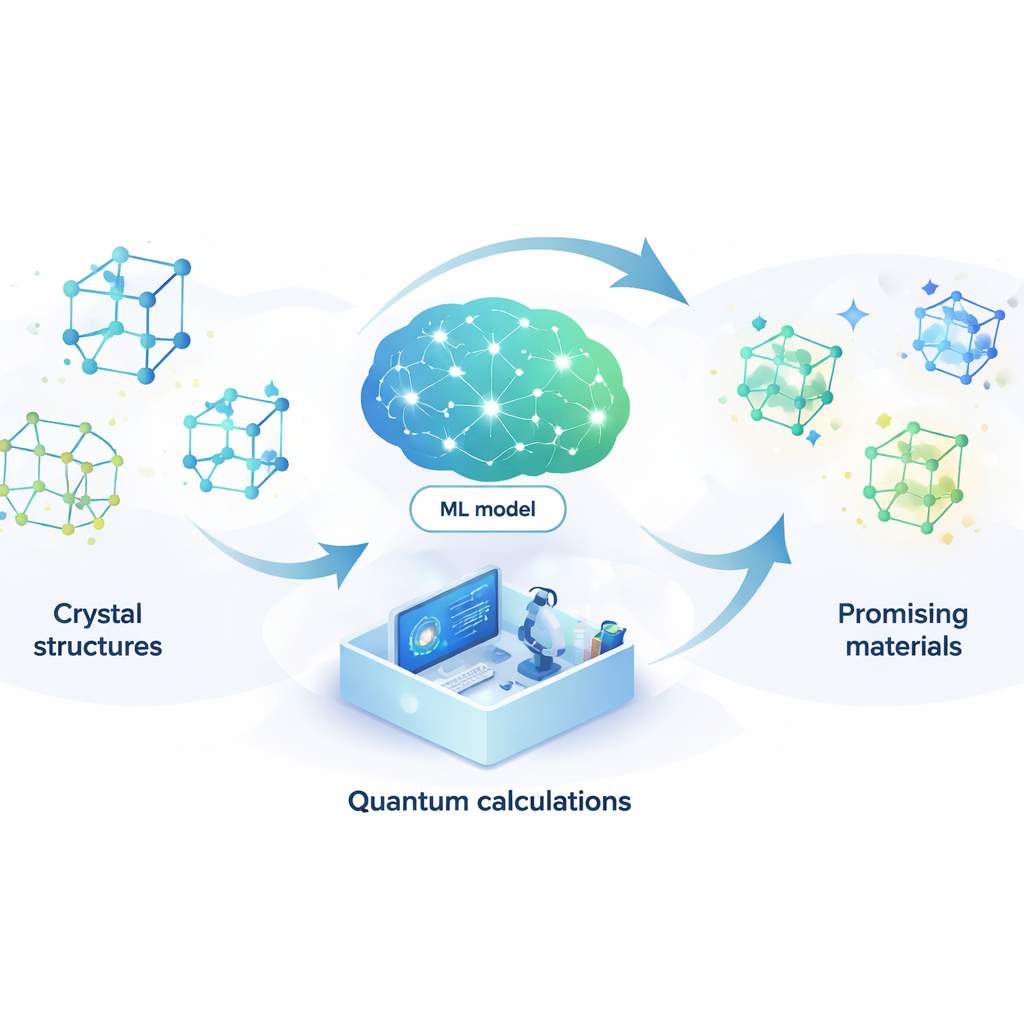
Permettere a un modello di apprendimento di sostituire la fisica quantistica
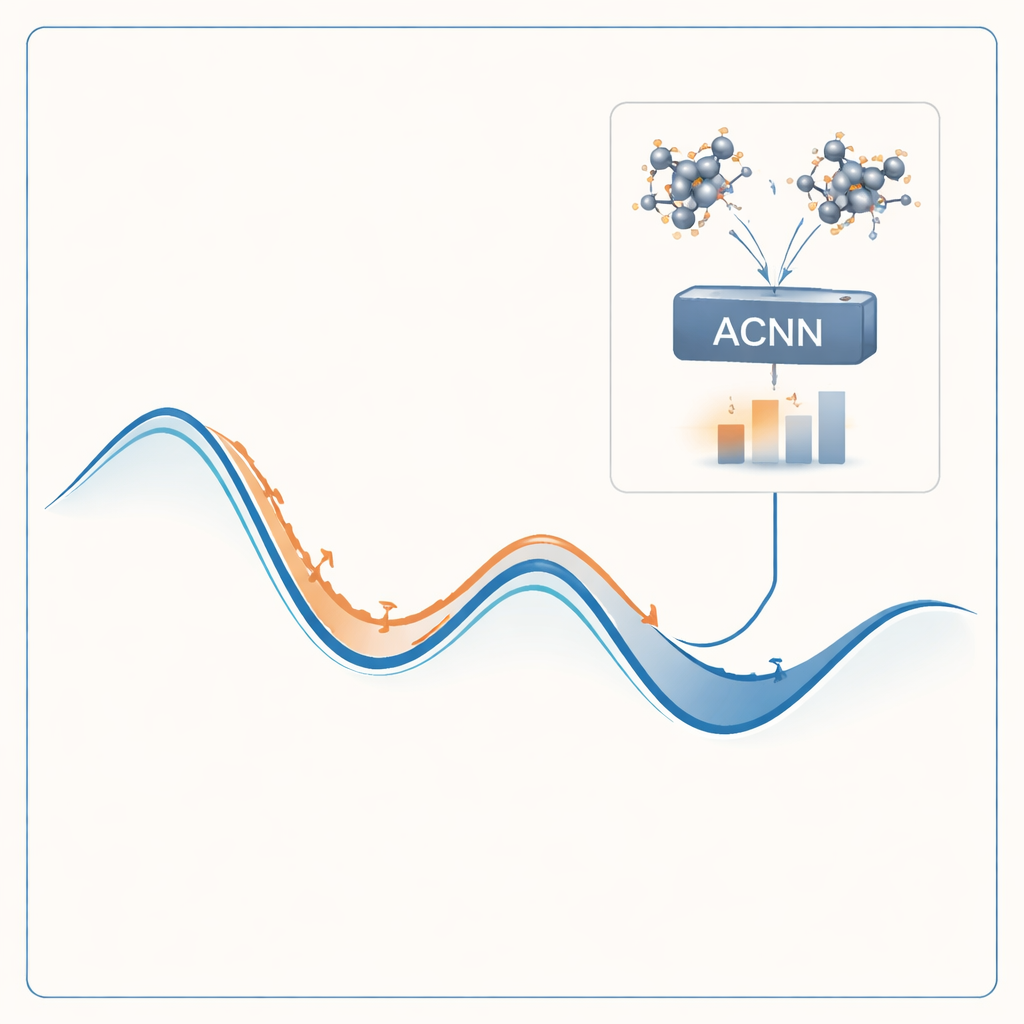
Per affrontare questo problema, gli autori costruiscono un modello di machine learning in grado di imitare i risultati di costosi calcoli quantistici a una frazione minima del costo. Il loro modello, chiamato rete neurale accoppiata con attenzione (ACNN), impara come l’energia di un materiale dipenda dalle posizioni e dai tipi dei suoi atomi. Una volta addestrato, può stimare molto rapidamente se una struttura cristallina proposta è probabile che sia stabile o meno e quali forze agiscono su ciascun atomo. Fondamentale è che il modello è progettato per rispettare requisiti fisici di base, come il fatto che traslare o ruotare l’intero cristallo non dovrebbe cambiare la sua energia totale.
Un ciclo di scoperta dei materiali che si auto-migliora
Piuttosto che addestrare il modello una sola volta sperando che funzioni ovunque, gli autori lo incapsulano in un ciclo auto-ottimizzante. Il processo inizia con un piccolo insieme di strutture cristalline casuali, che vengono valutate con calcoli quantomeccanici completi e usate per addestrare una ACNN iniziale. Questo modello viene poi utilizzato per rilassare milioni di strutture di prova, trovando rapidamente minimi locali di energia — fasi candidate stabili o quasi stabili. Il flusso di lavoro segnala automaticamente due tipi di strutture particolarmente preziose: quelle che appaiono molto stabili e quelle che sembrano non fisiche o sospette. Solo questi casi selezionati vengono rimandati al costoso risolutore quantistico, e i nuovi risultati vengono reintegrati nel modello per un nuovo addestramento. Nel corso di molte iterazioni, il modello diventa progressivamente più accurato nelle regioni dello spazio delle strutture che contano di più.
Mettere alla prova il metodo
Il gruppo dimostra il proprio approccio su due sistemi impegnativi. Il primo è una miscela ad alta pressione di magnesio, calcio e idrogeno, una famiglia di composti di grande interesse per la superconduttività ad alta temperatura. Esplorando quasi sei milioni di strutture di prova, il flusso di lavoro scopre una nuova fase stabile, MgCa₃H₂₃, e diverse strutture “gabbia” ricche di idrogeno strettamente correlate. I calcoli suggeriscono che alcune di queste potrebbero essere superconduttive a temperature superiori al punto di ebollizione dell’azoto liquido sotto pressioni estreme. Il secondo test riguarda un sistema a quattro elementi contenente berillio, fosforo, azoto e ossigeno, scelto per il suo potenziale nel contenere cristalli che convertano efficientemente la luce laser in lunghezze d’onda nell’estremo ultravioletto. Qui il metodo rilassa più di nove milioni di strutture e identifica tre fasi termodinamicamente stabili con gap di banda molto ampi e caratteristiche ottiche promettenti.
Dalla forza bruta alla scoperta guidata
In entrambi gli esempi, il flusso di lavoro automatizzato raggiunge accelerazioni di circa diecimila volte rispetto all’uso esclusivo di calcoli quantistici, pur individuando in modo affidabile le strutture meritevoli di uno studio più approfondito. Per un non specialista, il messaggio chiave è che gran parte della scoperta dei materiali può ora essere gestita da un sistema di apprendimento che impara dove è incerto e richiede calcoli mirati ad alta precisione solo quando necessario. Questo tipo di ricerca auto-correggente assistita dall’IA apre la porta all’esplorazione di miscele di elementi molto più complesse di quanto fosse precedentemente fattibile, aumentando le probabilità di scoprire nuovi superconduttori, cristalli ottici e altri materiali funzionali che sosterranno le tecnologie di nuova generazione.
Citazione: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Parole chiave: scoperta dei materiali, potenziali di machine learning, predizione della struttura cristallina, idruro superconduttori, cristalli ottici non lineari